Vibrational Frequencies calculated at mPW1PW91/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3832 |
3634 |
17.92 |
|
|
|
| 2 |
A |
3817 |
3619 |
13.60 |
|
|
|
| 3 |
A |
3165 |
3001 |
29.93 |
|
|
|
| 4 |
A |
3143 |
2980 |
33.18 |
|
|
|
| 5 |
A |
3137 |
2975 |
29.01 |
|
|
|
| 6 |
A |
3086 |
2927 |
27.48 |
|
|
|
| 7 |
A |
3081 |
2922 |
25.10 |
|
|
|
| 8 |
A |
3069 |
2910 |
21.17 |
|
|
|
| 9 |
A |
3007 |
2852 |
97.00 |
|
|
|
| 10 |
A |
2994 |
2839 |
32.66 |
|
|
|
| 11 |
A |
1563 |
1483 |
3.62 |
|
|
|
| 12 |
A |
1532 |
1453 |
4.39 |
|
|
|
| 13 |
A |
1527 |
1448 |
4.88 |
|
|
|
| 14 |
A |
1514 |
1436 |
1.96 |
|
|
|
| 15 |
A |
1489 |
1412 |
15.08 |
|
|
|
| 16 |
A |
1460 |
1385 |
16.84 |
|
|
|
| 17 |
A |
1438 |
1364 |
13.07 |
|
|
|
| 18 |
A |
1407 |
1334 |
6.97 |
|
|
|
| 19 |
A |
1348 |
1278 |
38.51 |
|
|
|
| 20 |
A |
1341 |
1272 |
3.39 |
|
|
|
| 21 |
A |
1314 |
1246 |
34.82 |
|
|
|
| 22 |
A |
1271 |
1205 |
59.62 |
|
|
|
| 23 |
A |
1235 |
1171 |
4.05 |
|
|
|
| 24 |
A |
1182 |
1121 |
37.45 |
|
|
|
| 25 |
A |
1157 |
1097 |
38.15 |
|
|
|
| 26 |
A |
1117 |
1059 |
29.50 |
|
|
|
| 27 |
A |
1094 |
1038 |
68.53 |
|
|
|
| 28 |
A |
1053 |
999 |
9.86 |
|
|
|
| 29 |
A |
1003 |
951 |
7.23 |
|
|
|
| 30 |
A |
964 |
915 |
7.96 |
|
|
|
| 31 |
A |
877 |
832 |
14.94 |
|
|
|
| 32 |
A |
813 |
771 |
3.98 |
|
|
|
| 33 |
A |
507 |
481 |
19.08 |
|
|
|
| 34 |
A |
485 |
460 |
5.99 |
|
|
|
| 35 |
A |
397 |
377 |
7.76 |
|
|
|
| 36 |
A |
341 |
323 |
8.88 |
|
|
|
| 37 |
A |
310 |
294 |
133.62 |
|
|
|
| 38 |
A |
283 |
268 |
105.51 |
|
|
|
| 39 |
A |
252 |
239 |
1.01 |
|
|
|
| 40 |
A |
180 |
171 |
3.77 |
|
|
|
| 41 |
A |
118 |
112 |
7.04 |
|
|
|
| 42 |
A |
94 |
89 |
6.89 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31498.8 cm
-1
Scaled (by 0.9483) Zero Point Vibrational Energy (zpe) 29870.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
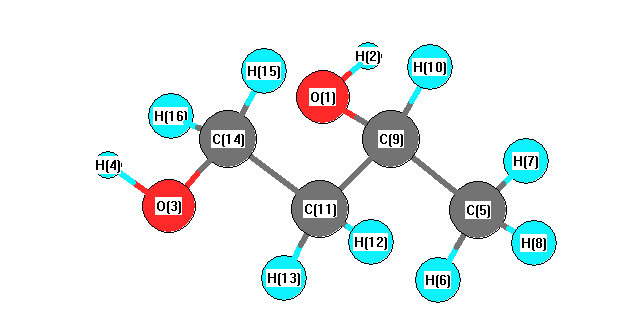
Charges from optimized geometry at mPW1PW91/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.639 |
|
|
|
| 2 |
H |
0.405 |
|
|
|
| 3 |
O |
-0.641 |
|
|
|
| 4 |
H |
0.407 |
|
|
|
| 5 |
C |
-0.534 |
|
|
|
| 6 |
H |
0.184 |
|
|
|
| 7 |
H |
0.159 |
|
|
|
| 8 |
H |
0.173 |
|
|
|
| 9 |
C |
0.089 |
|
|
|
| 10 |
H |
0.140 |
|
|
|
| 11 |
C |
-0.329 |
|
|
|
| 12 |
H |
0.175 |
|
|
|
| 13 |
H |
0.183 |
|
|
|
| 14 |
C |
-0.079 |
|
|
|
| 15 |
H |
0.135 |
|
|
|
| 16 |
H |
0.171 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.162 |
0.884 |
1.394 |
2.019 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.985 |
-2.792 |
-0.188 |
| y |
-2.792 |
-38.018 |
0.942 |
| z |
-0.188 |
0.942 |
-38.276 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.162 |
-2.792 |
-0.188 |
| y |
-2.792 |
-3.388 |
0.942 |
| z |
-0.188 |
0.942 |
-3.775 |
|
| Polar |
|---|
| 3z2-r2 | -7.550 |
|---|
| x2-y2 | 7.033 |
|---|
| xy | -2.792 |
|---|
| xz | -0.188 |
|---|
| yz | 0.942 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
222.187 |
| (<r2>)1/2 |
14.906 |