Jump to
S1C2
Energy calculated at mPW1PW91/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -251.871503 |
| Energy at 298.15K | -251.885856 |
| Nuclear repulsion energy | 259.927580 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3583 |
3421 |
0.25 |
|
|
|
| 2 |
A' |
3116 |
2975 |
61.68 |
|
|
|
| 3 |
A' |
3100 |
2959 |
31.24 |
|
|
|
| 4 |
A' |
3092 |
2952 |
34.49 |
|
|
|
| 5 |
A' |
3059 |
2920 |
16.34 |
|
|
|
| 6 |
A' |
3041 |
2903 |
27.15 |
|
|
|
| 7 |
A' |
2928 |
2796 |
132.54 |
|
|
|
| 8 |
A' |
1509 |
1440 |
2.93 |
|
|
|
| 9 |
A' |
1493 |
1425 |
6.34 |
|
|
|
| 10 |
A' |
1482 |
1415 |
7.20 |
|
|
|
| 11 |
A' |
1432 |
1367 |
3.21 |
|
|
|
| 12 |
A' |
1385 |
1322 |
0.14 |
|
|
|
| 13 |
A' |
1323 |
1263 |
0.85 |
|
|
|
| 14 |
A' |
1293 |
1235 |
3.78 |
|
|
|
| 15 |
A' |
1173 |
1120 |
5.39 |
|
|
|
| 16 |
A' |
1074 |
1026 |
3.61 |
|
|
|
| 17 |
A' |
1064 |
1016 |
7.92 |
|
|
|
| 18 |
A' |
928 |
886 |
1.30 |
|
|
|
| 19 |
A' |
875 |
836 |
14.34 |
|
|
|
| 20 |
A' |
842 |
804 |
0.29 |
|
|
|
| 21 |
A' |
775 |
740 |
75.92 |
|
|
|
| 22 |
A' |
538 |
513 |
19.45 |
|
|
|
| 23 |
A' |
433 |
414 |
6.33 |
|
|
|
| 24 |
A' |
393 |
375 |
1.29 |
|
|
|
| 25 |
A' |
244 |
233 |
0.97 |
|
|
|
| 26 |
A" |
3111 |
2970 |
21.74 |
|
|
|
| 27 |
A" |
3091 |
2951 |
75.42 |
|
|
|
| 28 |
A" |
3059 |
2921 |
20.36 |
|
|
|
| 29 |
A" |
2923 |
2791 |
29.08 |
|
|
|
| 30 |
A" |
1500 |
1432 |
1.53 |
|
|
|
| 31 |
A" |
1490 |
1423 |
0.64 |
|
|
|
| 32 |
A" |
1472 |
1405 |
3.01 |
|
|
|
| 33 |
A" |
1386 |
1323 |
0.31 |
|
|
|
| 34 |
A" |
1367 |
1305 |
23.38 |
|
|
|
| 35 |
A" |
1350 |
1288 |
10.57 |
|
|
|
| 36 |
A" |
1300 |
1241 |
2.66 |
|
|
|
| 37 |
A" |
1202 |
1148 |
11.57 |
|
|
|
| 38 |
A" |
1170 |
1117 |
3.16 |
|
|
|
| 39 |
A" |
1160 |
1107 |
7.70 |
|
|
|
| 40 |
A" |
1078 |
1030 |
2.02 |
|
|
|
| 41 |
A" |
981 |
936 |
0.70 |
|
|
|
| 42 |
A" |
899 |
859 |
0.35 |
|
|
|
| 43 |
A" |
821 |
784 |
0.28 |
|
|
|
| 44 |
A" |
449 |
429 |
0.76 |
|
|
|
| 45 |
A" |
244 |
233 |
0.43 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35113.1 cm
-1
Scaled (by 0.9547) Zero Point Vibrational Energy (zpe) 33522.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31G(2df,p)
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.636 |
1.323 |
0.000 |
| H2 |
0.577 |
2.416 |
0.000 |
| H3 |
1.704 |
1.067 |
0.000 |
| C4 |
-0.015 |
0.747 |
1.256 |
| C5 |
-0.015 |
0.747 |
-1.256 |
| C6 |
-0.015 |
-0.776 |
-1.205 |
| C7 |
-0.015 |
-0.776 |
1.205 |
| N8 |
-0.694 |
-1.224 |
0.000 |
| H9 |
-0.766 |
-2.232 |
0.000 |
| H10 |
0.507 |
1.090 |
2.155 |
| H11 |
0.507 |
1.090 |
-2.155 |
| H12 |
-1.052 |
1.094 |
1.325 |
| H13 |
-1.052 |
1.094 |
-1.325 |
| H14 |
1.032 |
-1.132 |
-1.257 |
| H15 |
1.032 |
-1.132 |
1.257 |
| H16 |
-0.538 |
-1.187 |
-2.074 |
| H17 |
-0.538 |
-1.187 |
2.074 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
C4 |
C5 |
C6 |
C7 |
N8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
| C1 | | 1.0943 | 1.0980 | 1.5277 | 1.5277 | 2.5067 | 2.5067 | 2.8735 | 3.8219 | 2.1714 | 2.1714 | 2.1588 | 2.1588 | 2.7866 | 2.7866 | 3.4611 | 3.4611 |
H2 | 1.0943 | | 1.7575 | 2.1709 | 2.1709 | 3.4629 | 3.4629 | 3.8553 | 4.8382 | 2.5312 | 2.5312 | 2.4816 | 2.4816 | 3.7915 | 3.7915 | 4.3037 | 4.3037 | H3 | 1.0980 | 1.7575 | | 2.1532 | 2.1532 | 2.7940 | 2.7940 | 3.3167 | 4.1216 | 2.4653 | 2.4653 | 3.0585 | 3.0585 | 2.6208 | 2.6208 | 3.7959 | 3.7959 | C4 | 1.5277 | 2.1709 | 2.1532 | | 2.5123 | 2.8943 | 1.5245 | 2.4340 | 3.3195 | 1.0944 | 3.4678 | 1.0956 | 2.8036 | 3.3084 | 2.1516 | 3.8864 | 2.1637 | C5 | 1.5277 | 2.1709 | 2.1532 | 2.5123 | | 1.5245 | 2.8943 | 2.4340 | 3.3195 | 3.4678 | 1.0944 | 2.8036 | 1.0956 | 2.1516 | 3.3084 | 2.1637 | 3.8864 | C6 | 2.5067 | 3.4629 | 2.7940 | 2.8943 | 1.5245 | | 2.4093 | 1.4534 | 2.0333 | 3.8784 | 2.1583 | 3.3128 | 2.1419 | 1.1077 | 2.6990 | 1.0943 | 3.3455 | C7 | 2.5067 | 3.4629 | 2.7940 | 1.5245 | 2.8943 | 2.4093 | | 1.4534 | 2.0333 | 2.1583 | 3.8784 | 2.1419 | 3.3128 | 2.6990 | 1.1077 | 3.3455 | 1.0943 | N8 | 2.8735 | 3.8553 | 3.3167 | 2.4340 | 2.4340 | 1.4534 | 1.4534 | | 1.0110 | 3.3822 | 3.3822 | 2.6940 | 2.6940 | 2.1376 | 2.1376 | 2.0804 | 2.0804 | H9 | 3.8219 | 4.8382 | 4.1216 | 3.3195 | 3.3195 | 2.0333 | 2.0333 | 1.0110 | | 4.1593 | 4.1593 | 3.5921 | 3.5921 | 2.4544 | 2.4544 | 2.3341 | 2.3341 | H10 | 2.1714 | 2.5312 | 2.4653 | 1.0944 | 3.4678 | 3.8784 | 2.1583 | 3.3822 | 4.1593 | | 4.3099 | 1.7659 | 3.8136 | 4.1055 | 2.4534 | 4.9151 | 2.5059 | H11 | 2.1714 | 2.5312 | 2.4653 | 3.4678 | 1.0944 | 2.1583 | 3.8784 | 3.3822 | 4.1593 | 4.3099 | | 3.8136 | 1.7659 | 2.4534 | 4.1055 | 2.5059 | 4.9151 | H12 | 2.1588 | 2.4816 | 3.0585 | 1.0956 | 2.8036 | 3.3128 | 2.1419 | 2.6940 | 3.5921 | 1.7659 | 3.8136 | | 2.6509 | 3.9963 | 3.0505 | 4.1258 | 2.4546 | H13 | 2.1588 | 2.4816 | 3.0585 | 2.8036 | 1.0956 | 2.1419 | 3.3128 | 2.6940 | 3.5921 | 3.8136 | 1.7659 | 2.6509 | | 3.0505 | 3.9963 | 2.4546 | 4.1258 | H14 | 2.7866 | 3.7915 | 2.6208 | 3.3084 | 2.1516 | 1.1077 | 2.6990 | 2.1376 | 2.4544 | 4.1055 | 2.4534 | 3.9963 | 3.0505 | | 2.5141 | 1.7711 | 3.6833 | H15 | 2.7866 | 3.7915 | 2.6208 | 2.1516 | 3.3084 | 2.6990 | 1.1077 | 2.1376 | 2.4544 | 2.4534 | 4.1055 | 3.0505 | 3.9963 | 2.5141 | | 3.6833 | 1.7711 | H16 | 3.4611 | 4.3037 | 3.7959 | 3.8864 | 2.1637 | 1.0943 | 3.3455 | 2.0804 | 2.3341 | 4.9151 | 2.5059 | 4.1258 | 2.4546 | 1.7711 | 3.6833 | | 4.1484 | H17 | 3.4611 | 4.3037 | 3.7959 | 2.1637 | 3.8864 | 3.3455 | 1.0943 | 2.0804 | 2.3341 | 2.5059 | 4.9151 | 2.4546 | 4.1258 | 3.6833 | 1.7711 | 4.1484 | |
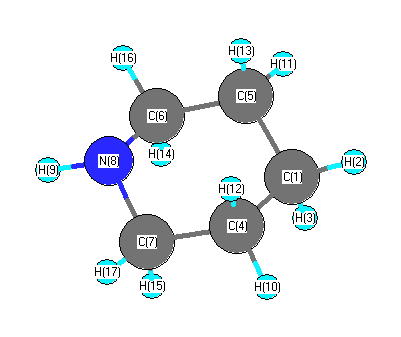 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C4 |
C7 |
110.425 |
|
C1 |
C4 |
H10 |
110.726 |
| C1 |
C4 |
H12 |
109.654 |
|
C1 |
C5 |
C6 |
110.425 |
| C1 |
C5 |
H11 |
110.726 |
|
C1 |
C5 |
H13 |
109.654 |
| H2 |
C1 |
H3 |
106.582 |
|
H2 |
C1 |
C4 |
110.687 |
| H2 |
C1 |
C5 |
110.687 |
|
H3 |
C1 |
C4 |
109.078 |
| H3 |
C1 |
C5 |
109.078 |
|
C4 |
C1 |
C5 |
110.620 |
| C4 |
C7 |
N8 |
109.621 |
|
C4 |
C7 |
H15 |
108.621 |
| C4 |
C7 |
H17 |
110.350 |
|
C5 |
C6 |
N8 |
109.621 |
| C5 |
C6 |
H14 |
108.621 |
|
C5 |
C6 |
H16 |
110.350 |
| C6 |
C5 |
H11 |
109.914 |
|
C6 |
C5 |
H13 |
108.561 |
| C6 |
N8 |
C7 |
111.969 |
|
C6 |
N8 |
H9 |
109.894 |
| C7 |
C4 |
H10 |
109.914 |
|
C7 |
C4 |
H12 |
108.561 |
| C7 |
N8 |
H9 |
109.894 |
|
N8 |
C6 |
H14 |
112.461 |
| N8 |
C6 |
H16 |
108.671 |
|
N8 |
C7 |
H15 |
112.461 |
| N8 |
C7 |
H17 |
108.671 |
|
H10 |
C4 |
H12 |
107.487 |
| H11 |
C5 |
H13 |
107.487 |
|
H14 |
C6 |
H16 |
107.089 |
| H15 |
C7 |
H17 |
107.089 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.249 |
|
|
|
| 2 |
H |
0.122 |
|
|
|
| 3 |
H |
0.120 |
|
|
|
| 4 |
C |
-0.231 |
|
|
|
| 5 |
C |
-0.231 |
|
|
|
| 6 |
C |
-0.186 |
|
|
|
| 7 |
C |
-0.186 |
|
|
|
| 8 |
N |
-0.340 |
|
|
|
| 9 |
H |
0.240 |
|
|
|
| 10 |
H |
0.119 |
|
|
|
| 11 |
H |
0.119 |
|
|
|
| 12 |
H |
0.131 |
|
|
|
| 13 |
H |
0.131 |
|
|
|
| 14 |
H |
0.097 |
|
|
|
| 15 |
H |
0.097 |
|
|
|
| 16 |
H |
0.124 |
|
|
|
| 17 |
H |
0.124 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.621 |
-0.465 |
0.000 |
0.776 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.263 |
-0.067 |
0.000 |
| y |
-0.067 |
-35.910 |
0.000 |
| z |
0.000 |
0.000 |
-37.479 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.568 |
-0.067 |
0.000 |
| y |
-0.067 |
2.961 |
0.000 |
| z |
0.000 |
0.000 |
0.607 |
|
| Polar |
|---|
| 3z2-r2 | 1.214 |
|---|
| x2-y2 | -4.353 |
|---|
| xy | -0.067 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.963 |
0.324 |
0.000 |
| y |
0.324 |
9.014 |
0.000 |
| z |
0.000 |
0.000 |
9.447 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |
Jump to
S1C1
Energy calculated at mPW1PW91/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -251.870769 |
| Energy at 298.15K | -251.885066 |
| Nuclear repulsion energy | 259.413511 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3546 |
3385 |
0.21 |
|
|
|
| 2 |
A' |
3104 |
2963 |
77.08 |
|
|
|
| 3 |
A' |
3097 |
2957 |
28.53 |
|
|
|
| 4 |
A' |
3093 |
2953 |
35.08 |
|
|
|
| 5 |
A' |
3042 |
2904 |
41.98 |
|
|
|
| 6 |
A' |
3037 |
2900 |
48.43 |
|
|
|
| 7 |
A' |
3035 |
2898 |
6.72 |
|
|
|
| 8 |
A' |
1501 |
1433 |
2.22 |
|
|
|
| 9 |
A' |
1484 |
1417 |
10.21 |
|
|
|
| 10 |
A' |
1482 |
1415 |
2.84 |
|
|
|
| 11 |
A' |
1403 |
1339 |
4.81 |
|
|
|
| 12 |
A' |
1385 |
1323 |
1.47 |
|
|
|
| 13 |
A' |
1347 |
1286 |
0.09 |
|
|
|
| 14 |
A' |
1286 |
1228 |
8.56 |
|
|
|
| 15 |
A' |
1196 |
1141 |
2.80 |
|
|
|
| 16 |
A' |
1062 |
1014 |
0.88 |
|
|
|
| 17 |
A' |
1025 |
979 |
8.51 |
|
|
|
| 18 |
A' |
932 |
890 |
5.10 |
|
|
|
| 19 |
A' |
869 |
830 |
39.67 |
|
|
|
| 20 |
A' |
834 |
796 |
0.12 |
|
|
|
| 21 |
A' |
771 |
736 |
93.44 |
|
|
|
| 22 |
A' |
544 |
519 |
1.74 |
|
|
|
| 23 |
A' |
435 |
415 |
1.02 |
|
|
|
| 24 |
A' |
382 |
365 |
5.20 |
|
|
|
| 25 |
A' |
237 |
226 |
4.66 |
|
|
|
| 26 |
A" |
3099 |
2959 |
20.39 |
|
|
|
| 27 |
A" |
3092 |
2952 |
77.96 |
|
|
|
| 28 |
A" |
3038 |
2900 |
3.28 |
|
|
|
| 29 |
A" |
3031 |
2894 |
41.68 |
|
|
|
| 30 |
A" |
1502 |
1434 |
8.30 |
|
|
|
| 31 |
A" |
1480 |
1413 |
3.05 |
|
|
|
| 32 |
A" |
1470 |
1404 |
2.57 |
|
|
|
| 33 |
A" |
1386 |
1323 |
1.74 |
|
|
|
| 34 |
A" |
1374 |
1312 |
2.40 |
|
|
|
| 35 |
A" |
1349 |
1287 |
0.84 |
|
|
|
| 36 |
A" |
1304 |
1245 |
0.80 |
|
|
|
| 37 |
A" |
1229 |
1173 |
17.03 |
|
|
|
| 38 |
A" |
1152 |
1100 |
14.72 |
|
|
|
| 39 |
A" |
1123 |
1072 |
1.09 |
|
|
|
| 40 |
A" |
1077 |
1028 |
2.40 |
|
|
|
| 41 |
A" |
950 |
907 |
4.15 |
|
|
|
| 42 |
A" |
891 |
851 |
0.24 |
|
|
|
| 43 |
A" |
807 |
770 |
0.06 |
|
|
|
| 44 |
A" |
454 |
433 |
0.77 |
|
|
|
| 45 |
A" |
231 |
220 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35082.6 cm
-1
Scaled (by 0.9547) Zero Point Vibrational Energy (zpe) 33493.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31G(2df,p)
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.641 |
1.319 |
0.000 |
| H2 |
0.583 |
2.412 |
0.000 |
| H3 |
1.709 |
1.064 |
0.000 |
| C4 |
-0.009 |
0.743 |
1.257 |
| C5 |
-0.009 |
0.743 |
-1.257 |
| C6 |
-0.009 |
-0.787 |
-1.207 |
| C7 |
-0.009 |
-0.787 |
1.207 |
| N8 |
-0.607 |
-1.342 |
0.000 |
| H9 |
-1.599 |
-1.135 |
0.000 |
| H10 |
0.508 |
1.092 |
2.157 |
| H11 |
0.508 |
1.092 |
-2.157 |
| H12 |
-1.045 |
1.098 |
1.330 |
| H13 |
-1.045 |
1.098 |
-1.330 |
| H14 |
1.027 |
-1.149 |
-1.258 |
| H15 |
1.027 |
-1.149 |
1.258 |
| H16 |
-0.528 |
-1.204 |
-2.075 |
| H17 |
-0.528 |
-1.204 |
2.075 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
C4 |
C5 |
C6 |
C7 |
N8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
| C1 | | 1.0943 | 1.0980 | 1.5283 | 1.5283 | 2.5128 | 2.5128 | 2.9392 | 3.3231 | 2.1731 | 2.1731 | 2.1592 | 2.1592 | 2.7965 | 2.7965 | 3.4698 | 3.4698 |
H2 | 1.0943 | | 1.7564 | 2.1720 | 2.1720 | 3.4698 | 3.4698 | 3.9380 | 4.1647 | 2.5298 | 2.5298 | 2.4792 | 2.4792 | 3.8021 | 3.8021 | 4.3147 | 4.3147 | H3 | 1.0980 | 1.7564 | | 2.1531 | 2.1531 | 2.7989 | 2.7989 | 3.3393 | 3.9725 | 2.4691 | 2.4691 | 3.0589 | 3.0589 | 2.6349 | 2.6349 | 3.8018 | 3.8018 | C4 | 1.5283 | 2.1720 | 2.1531 | | 2.5144 | 2.9000 | 1.5304 | 2.5067 | 2.7632 | 1.0954 | 3.4710 | 1.0980 | 2.8098 | 3.3130 | 2.1563 | 3.8942 | 2.1744 | C5 | 1.5283 | 2.1720 | 2.1531 | 2.5144 | | 1.5304 | 2.9000 | 2.5067 | 2.7632 | 3.4710 | 1.0954 | 2.8098 | 1.0980 | 2.1563 | 3.3130 | 2.1744 | 3.8942 | C6 | 2.5128 | 3.4698 | 2.7989 | 2.9000 | 1.5304 | | 2.4132 | 1.4564 | 2.0263 | 3.8878 | 2.1688 | 3.3262 | 2.1549 | 1.0985 | 2.6979 | 1.0944 | 3.3487 | C7 | 2.5128 | 3.4698 | 2.7989 | 1.5304 | 2.9000 | 2.4132 | | 1.4564 | 2.0263 | 2.1688 | 3.8878 | 2.1549 | 3.3262 | 2.6979 | 1.0985 | 3.3487 | 1.0944 | N8 | 2.9392 | 3.9380 | 3.3393 | 2.5067 | 2.5067 | 1.4564 | 1.4564 | | 1.0140 | 3.4386 | 3.4386 | 2.8139 | 2.8139 | 2.0709 | 2.0709 | 2.0814 | 2.0814 | H9 | 3.3231 | 4.1647 | 3.9725 | 2.7632 | 2.7632 | 2.0263 | 2.0263 | 1.0140 | | 3.7493 | 3.7493 | 2.6581 | 2.6581 | 2.9120 | 2.9120 | 2.3367 | 2.3367 | H10 | 2.1731 | 2.5298 | 2.4691 | 1.0954 | 3.4710 | 3.8878 | 2.1688 | 3.4386 | 3.7493 | | 4.3144 | 1.7598 | 3.8177 | 4.1177 | 2.4698 | 4.9256 | 2.5208 | H11 | 2.1731 | 2.5298 | 2.4691 | 3.4710 | 1.0954 | 2.1688 | 3.8878 | 3.4386 | 3.7493 | 4.3144 | | 3.8177 | 1.7598 | 2.4698 | 4.1177 | 2.5208 | 4.9256 | H12 | 2.1592 | 2.4792 | 3.0589 | 1.0980 | 2.8098 | 3.3262 | 2.1549 | 2.8139 | 2.6581 | 1.7598 | 3.8177 | | 2.6604 | 4.0052 | 3.0574 | 4.1433 | 2.4748 | H13 | 2.1592 | 2.4792 | 3.0589 | 2.8098 | 1.0980 | 2.1549 | 3.3262 | 2.8139 | 2.6581 | 3.8177 | 1.7598 | 2.6604 | | 3.0574 | 4.0052 | 2.4748 | 4.1433 | H14 | 2.7965 | 3.8021 | 2.6349 | 3.3130 | 2.1563 | 1.0985 | 2.6979 | 2.0709 | 2.9120 | 4.1177 | 2.4698 | 4.0052 | 3.0574 | | 2.5161 | 1.7572 | 3.6785 | H15 | 2.7965 | 3.8021 | 2.6349 | 2.1563 | 3.3130 | 2.6979 | 1.0985 | 2.0709 | 2.9120 | 2.4698 | 4.1177 | 3.0574 | 4.0052 | 2.5161 | | 3.6785 | 1.7572 | H16 | 3.4698 | 4.3147 | 3.8018 | 3.8942 | 2.1744 | 1.0944 | 3.3487 | 2.0814 | 2.3367 | 4.9256 | 2.5208 | 4.1433 | 2.4748 | 1.7572 | 3.6785 | | 4.1507 | H17 | 3.4698 | 4.3147 | 3.8018 | 2.1744 | 3.8942 | 3.3487 | 1.0944 | 2.0814 | 2.3367 | 2.5208 | 4.9256 | 2.4748 | 4.1433 | 3.6785 | 1.7572 | 4.1507 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C4 |
C7 |
110.474 |
|
C1 |
C4 |
H10 |
110.762 |
| C1 |
C4 |
H12 |
109.510 |
|
C1 |
C5 |
C6 |
110.474 |
| C1 |
C5 |
H11 |
110.762 |
|
C1 |
C5 |
H13 |
109.510 |
| H2 |
C1 |
H3 |
106.478 |
|
H2 |
C1 |
C4 |
110.739 |
| H2 |
C1 |
C5 |
110.739 |
|
H3 |
C1 |
C4 |
109.035 |
| H3 |
C1 |
C5 |
109.035 |
|
C4 |
C1 |
C5 |
110.697 |
| C4 |
C7 |
N8 |
114.102 |
|
C4 |
C7 |
H15 |
109.109 |
| C4 |
C7 |
H17 |
110.776 |
|
C5 |
C6 |
N8 |
114.102 |
| C5 |
C6 |
H14 |
109.109 |
|
C5 |
C6 |
H16 |
110.776 |
| C6 |
C5 |
H11 |
110.272 |
|
C6 |
C5 |
H13 |
109.030 |
| C6 |
N8 |
C7 |
111.887 |
|
C6 |
N8 |
H9 |
108.896 |
| C7 |
C4 |
H10 |
110.272 |
|
C7 |
C4 |
H12 |
109.030 |
| C7 |
N8 |
H9 |
108.896 |
|
N8 |
C6 |
H14 |
107.477 |
| N8 |
C6 |
H16 |
108.540 |
|
N8 |
C7 |
H15 |
107.477 |
| N8 |
C7 |
H17 |
108.540 |
|
H10 |
C4 |
H12 |
106.704 |
| H11 |
C5 |
H13 |
106.704 |
|
H14 |
C6 |
H16 |
106.515 |
| H15 |
C7 |
H17 |
106.515 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.243 |
|
|
|
| 2 |
H |
0.123 |
|
|
|
| 3 |
H |
0.123 |
|
|
|
| 4 |
C |
-0.251 |
|
|
|
| 5 |
C |
-0.251 |
|
|
|
| 6 |
C |
-0.191 |
|
|
|
| 7 |
C |
-0.191 |
|
|
|
| 8 |
N |
-0.331 |
|
|
|
| 9 |
H |
0.233 |
|
|
|
| 10 |
H |
0.121 |
|
|
|
| 11 |
H |
0.121 |
|
|
|
| 12 |
H |
0.116 |
|
|
|
| 13 |
H |
0.116 |
|
|
|
| 14 |
H |
0.127 |
|
|
|
| 15 |
H |
0.127 |
|
|
|
| 16 |
H |
0.126 |
|
|
|
| 17 |
H |
0.126 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.278 |
1.050 |
0.000 |
1.086 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.288 |
-0.291 |
0.000 |
| y |
-0.291 |
-42.529 |
0.000 |
| z |
0.000 |
0.000 |
-37.473 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.713 |
-0.291 |
0.000 |
| y |
-0.291 |
-5.648 |
0.000 |
| z |
0.000 |
0.000 |
1.935 |
|
| Polar |
|---|
| 3z2-r2 | 3.871 |
|---|
| x2-y2 | 6.241 |
|---|
| xy | -0.291 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.201 |
0.280 |
0.000 |
| y |
0.280 |
8.602 |
0.000 |
| z |
0.000 |
0.000 |
9.461 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |