Vibrational Frequencies calculated at mPW1PW91/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3633 |
3468 |
4.24 |
|
|
|
| 2 |
A |
3629 |
3464 |
0.94 |
|
|
|
| 3 |
A |
3544 |
3383 |
0.67 |
|
|
|
| 4 |
A |
3540 |
3379 |
5.16 |
|
|
|
| 5 |
A |
3141 |
2998 |
37.27 |
|
|
|
| 6 |
A |
3135 |
2993 |
33.21 |
|
|
|
| 7 |
A |
3115 |
2973 |
6.93 |
|
|
|
| 8 |
A |
3073 |
2934 |
51.72 |
|
|
|
| 9 |
A |
3068 |
2929 |
5.76 |
|
|
|
| 10 |
A |
3051 |
2912 |
26.43 |
|
|
|
| 11 |
A |
2957 |
2823 |
115.44 |
|
|
|
| 12 |
A |
2939 |
2806 |
56.98 |
|
|
|
| 13 |
A |
1659 |
1584 |
37.81 |
|
|
|
| 14 |
A |
1642 |
1568 |
45.76 |
|
|
|
| 15 |
A |
1513 |
1445 |
4.34 |
|
|
|
| 16 |
A |
1509 |
1441 |
7.26 |
|
|
|
| 17 |
A |
1500 |
1432 |
4.12 |
|
|
|
| 18 |
A |
1480 |
1413 |
1.67 |
|
|
|
| 19 |
A |
1437 |
1372 |
1.42 |
|
|
|
| 20 |
A |
1431 |
1366 |
10.36 |
|
|
|
| 21 |
A |
1411 |
1347 |
2.28 |
|
|
|
| 22 |
A |
1393 |
1330 |
12.14 |
|
|
|
| 23 |
A |
1345 |
1284 |
1.59 |
|
|
|
| 24 |
A |
1306 |
1247 |
0.98 |
|
|
|
| 25 |
A |
1292 |
1233 |
2.49 |
|
|
|
| 26 |
A |
1268 |
1211 |
1.77 |
|
|
|
| 27 |
A |
1199 |
1145 |
5.18 |
|
|
|
| 28 |
A |
1167 |
1114 |
6.55 |
|
|
|
| 29 |
A |
1120 |
1069 |
3.33 |
|
|
|
| 30 |
A |
1080 |
1031 |
5.42 |
|
|
|
| 31 |
A |
1076 |
1027 |
1.35 |
|
|
|
| 32 |
A |
1019 |
973 |
1.35 |
|
|
|
| 33 |
A |
989 |
944 |
23.22 |
|
|
|
| 34 |
A |
938 |
895 |
20.50 |
|
|
|
| 35 |
A |
917 |
875 |
52.68 |
|
|
|
| 36 |
A |
867 |
827 |
135.07 |
|
|
|
| 37 |
A |
824 |
787 |
21.40 |
|
|
|
| 38 |
A |
767 |
733 |
1.22 |
|
|
|
| 39 |
A |
624 |
595 |
16.80 |
|
|
|
| 40 |
A |
445 |
425 |
5.01 |
|
|
|
| 41 |
A |
380 |
363 |
3.36 |
|
|
|
| 42 |
A |
337 |
322 |
31.14 |
|
|
|
| 43 |
A |
278 |
265 |
32.41 |
|
|
|
| 44 |
A |
271 |
259 |
24.16 |
|
|
|
| 45 |
A |
235 |
224 |
10.27 |
|
|
|
| 46 |
A |
216 |
206 |
2.21 |
|
|
|
| 47 |
A |
146 |
140 |
5.26 |
|
|
|
| 48 |
A |
81 |
78 |
0.90 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36991.6 cm
-1
Scaled (by 0.9547) Zero Point Vibrational Energy (zpe) 35315.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
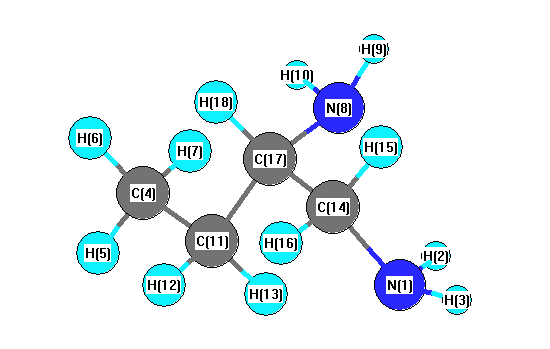
Charges from optimized geometry at mPW1PW91/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.562 |
|
|
|
| 2 |
H |
0.258 |
|
|
|
| 3 |
H |
0.246 |
|
|
|
| 4 |
C |
-0.470 |
|
|
|
| 5 |
H |
0.145 |
|
|
|
| 6 |
H |
0.136 |
|
|
|
| 7 |
H |
0.138 |
|
|
|
| 8 |
N |
-0.574 |
|
|
|
| 9 |
H |
0.247 |
|
|
|
| 10 |
H |
0.251 |
|
|
|
| 11 |
C |
-0.202 |
|
|
|
| 12 |
H |
0.123 |
|
|
|
| 13 |
H |
0.150 |
|
|
|
| 14 |
C |
-0.183 |
|
|
|
| 15 |
H |
0.093 |
|
|
|
| 16 |
H |
0.123 |
|
|
|
| 17 |
C |
-0.000 |
|
|
|
| 18 |
H |
0.082 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.052 |
1.341 |
1.123 |
1.750 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.372 |
0.414 |
0.740 |
| y |
0.414 |
-37.920 |
-0.357 |
| z |
0.740 |
-0.357 |
-40.117 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.647 |
0.414 |
0.740 |
| y |
0.414 |
-0.175 |
-0.357 |
| z |
0.740 |
-0.357 |
-3.471 |
|
| Polar |
|---|
| 3z2-r2 | -6.943 |
|---|
| x2-y2 | 2.548 |
|---|
| xy | 0.414 |
|---|
| xz | 0.740 |
|---|
| yz | -0.357 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.954 |
-0.019 |
0.100 |
| y |
-0.019 |
8.456 |
-0.071 |
| z |
0.100 |
-0.071 |
8.042 |
<r2> (average value of r
2) Å
2
| <r2> |
204.012 |
| (<r2>)1/2 |
14.283 |