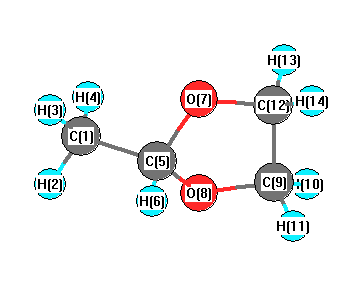
Vibrational Frequencies calculated at mPW1PW91/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3184 |
3039 |
22.60 |
|
|
|
| 2 |
A |
3179 |
3034 |
10.82 |
|
|
|
| 3 |
A |
3178 |
3034 |
26.01 |
|
|
|
| 4 |
A |
3171 |
3027 |
14.35 |
|
|
|
| 5 |
A |
3097 |
2957 |
5.81 |
|
|
|
| 6 |
A |
3091 |
2950 |
59.45 |
|
|
|
| 7 |
A |
3079 |
2939 |
51.10 |
|
|
|
| 8 |
A |
3071 |
2932 |
28.55 |
|
|
|
| 9 |
A |
1568 |
1497 |
0.18 |
|
|
|
| 10 |
A |
1562 |
1491 |
4.95 |
|
|
|
| 11 |
A |
1555 |
1484 |
3.00 |
|
|
|
| 12 |
A |
1550 |
1480 |
5.51 |
|
|
|
| 13 |
A |
1462 |
1395 |
24.47 |
|
|
|
| 14 |
A |
1428 |
1363 |
33.79 |
|
|
|
| 15 |
A |
1394 |
1330 |
0.47 |
|
|
|
| 16 |
A |
1387 |
1324 |
17.38 |
|
|
|
| 17 |
A |
1317 |
1257 |
9.36 |
|
|
|
| 18 |
A |
1257 |
1200 |
11.45 |
|
|
|
| 19 |
A |
1223 |
1168 |
0.36 |
|
|
|
| 20 |
A |
1167 |
1114 |
25.26 |
|
|
|
| 21 |
A |
1159 |
1106 |
88.52 |
|
|
|
| 22 |
A |
1132 |
1081 |
53.66 |
|
|
|
| 23 |
A |
1118 |
1068 |
52.08 |
|
|
|
| 24 |
A |
1047 |
999 |
15.38 |
|
|
|
| 25 |
A |
1016 |
970 |
31.55 |
|
|
|
| 26 |
A |
948 |
905 |
9.70 |
|
|
|
| 27 |
A |
923 |
881 |
1.45 |
|
|
|
| 28 |
A |
858 |
819 |
34.07 |
|
|
|
| 29 |
A |
833 |
795 |
6.85 |
|
|
|
| 30 |
A |
701 |
669 |
2.78 |
|
|
|
| 31 |
A |
629 |
600 |
5.11 |
|
|
|
| 32 |
A |
458 |
437 |
16.27 |
|
|
|
| 33 |
A |
349 |
333 |
2.55 |
|
|
|
| 34 |
A |
246 |
234 |
1.15 |
|
|
|
| 35 |
A |
229 |
218 |
0.42 |
|
|
|
| 36 |
A |
40 |
38 |
12.48 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 26800.5 cm
-1
Scaled (by 0.9546) Zero Point Vibrational Energy (zpe) 25583.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.631 |
|
|
|
| 2 |
H |
0.227 |
|
|
|
| 3 |
H |
0.224 |
|
|
|
| 4 |
H |
0.229 |
|
|
|
| 5 |
C |
0.228 |
|
|
|
| 6 |
H |
0.228 |
|
|
|
| 7 |
O |
-0.494 |
|
|
|
| 8 |
O |
-0.497 |
|
|
|
| 9 |
C |
-0.212 |
|
|
|
| 10 |
H |
0.235 |
|
|
|
| 11 |
H |
0.224 |
|
|
|
| 12 |
C |
-0.218 |
|
|
|
| 13 |
H |
0.222 |
|
|
|
| 14 |
H |
0.236 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.097 |
0.071 |
0.567 |
1.237 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.541 |
-0.230 |
-1.334 |
| y |
-0.230 |
-41.362 |
1.295 |
| z |
-1.334 |
1.295 |
-35.418 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.849 |
-0.230 |
-1.334 |
| y |
-0.230 |
-8.382 |
1.295 |
| z |
-1.334 |
1.295 |
0.534 |
|
| Polar |
|---|
| 3z2-r2 | 1.068 |
|---|
| x2-y2 | 10.821 |
|---|
| xy | -0.230 |
|---|
| xz | -1.334 |
|---|
| yz | 1.295 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
147.944 |
| (<r2>)1/2 |
12.163 |