Vibrational Frequencies calculated at mPW1PW91/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3169 |
3025 |
20.35 |
|
|
|
| 2 |
A |
3152 |
3009 |
16.41 |
|
|
|
| 3 |
A |
3144 |
3001 |
37.49 |
|
|
|
| 4 |
A |
3116 |
2975 |
4.10 |
|
|
|
| 5 |
A |
3114 |
2973 |
7.70 |
|
|
|
| 6 |
A |
3078 |
2939 |
19.23 |
|
|
|
| 7 |
A |
3071 |
2932 |
24.88 |
|
|
|
| 8 |
A |
1573 |
1502 |
7.90 |
|
|
|
| 9 |
A |
1571 |
1500 |
20.69 |
|
|
|
| 10 |
A |
1569 |
1498 |
7.77 |
|
|
|
| 11 |
A |
1537 |
1467 |
5.31 |
|
|
|
| 12 |
A |
1473 |
1406 |
12.02 |
|
|
|
| 13 |
A |
1467 |
1401 |
8.98 |
|
|
|
| 14 |
A |
1321 |
1261 |
21.42 |
|
|
|
| 15 |
A |
1318 |
1259 |
17.18 |
|
|
|
| 16 |
A |
1223 |
1168 |
12.90 |
|
|
|
| 17 |
A |
1128 |
1077 |
33.70 |
|
|
|
| 18 |
A |
1085 |
1036 |
1.20 |
|
|
|
| 19 |
A |
1009 |
963 |
7.63 |
|
|
|
| 20 |
A |
912 |
871 |
2.61 |
|
|
|
| 21 |
A |
664 |
634 |
2.44 |
|
|
|
| 22 |
A |
605 |
578 |
3.22 |
|
|
|
| 23 |
A |
460 |
439 |
3.03 |
|
|
|
| 24 |
A |
375 |
358 |
0.38 |
|
|
|
| 25 |
A |
294 |
280 |
1.45 |
|
|
|
| 26 |
A |
270 |
258 |
0.16 |
|
|
|
| 27 |
A |
162 |
154 |
0.34 |
|
|
|
| 28 |
A |
3186 |
3041 |
19.04 |
|
|
|
| 29 |
A |
3165 |
3021 |
12.67 |
|
|
|
| 30 |
A |
3155 |
3012 |
0.93 |
|
|
|
| 31 |
A |
3135 |
2992 |
0.42 |
|
|
|
| 32 |
A |
3067 |
2928 |
12.01 |
|
|
|
| 33 |
A |
1563 |
1492 |
7.71 |
|
|
|
| 34 |
A |
1557 |
1487 |
6.85 |
|
|
|
| 35 |
A |
1550 |
1480 |
0.20 |
|
|
|
| 36 |
A |
1456 |
1390 |
20.52 |
|
|
|
| 37 |
A |
1378 |
1315 |
0.61 |
|
|
|
| 38 |
A |
1314 |
1254 |
0.64 |
|
|
|
| 39 |
A |
1158 |
1106 |
3.41 |
|
|
|
| 40 |
A |
1067 |
1018 |
0.34 |
|
|
|
| 41 |
A |
994 |
949 |
0.81 |
|
|
|
| 42 |
A |
978 |
934 |
2.31 |
|
|
|
| 43 |
A |
817 |
780 |
7.78 |
|
|
|
| 44 |
A |
321 |
307 |
1.69 |
|
|
|
| 45 |
A |
255 |
243 |
0.00 |
|
|
|
| 46 |
A |
247 |
236 |
0.04 |
|
|
|
| 47 |
A |
78 |
75 |
2.28 |
|
|
|
| 48 |
A |
39 |
37 |
0.31 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36168.9 cm
-1
Scaled (by 0.9546) Zero Point Vibrational Energy (zpe) 34526.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
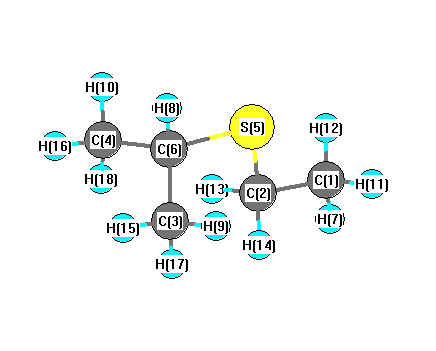
Charges from optimized geometry at mPW1PW91/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.633 |
|
|
|
| 2 |
C |
-0.689 |
|
|
|
| 3 |
C |
-0.601 |
|
|
|
| 4 |
C |
-0.601 |
|
|
|
| 5 |
S |
0.261 |
|
|
|
| 6 |
C |
-0.538 |
|
|
|
| 7 |
H |
0.224 |
|
|
|
| 8 |
H |
0.266 |
|
|
|
| 9 |
H |
0.234 |
|
|
|
| 10 |
H |
0.234 |
|
|
|
| 11 |
H |
0.235 |
|
|
|
| 12 |
H |
0.235 |
|
|
|
| 13 |
H |
0.250 |
|
|
|
| 14 |
H |
0.250 |
|
|
|
| 15 |
H |
0.220 |
|
|
|
| 16 |
H |
0.220 |
|
|
|
| 17 |
H |
0.217 |
|
|
|
| 18 |
H |
0.217 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.024 |
-2.025 |
0.000 |
2.025 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.338 |
0.362 |
0.000 |
| y |
0.362 |
-49.587 |
0.000 |
| z |
0.000 |
0.000 |
-47.825 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.368 |
0.362 |
0.000 |
| y |
0.362 |
-4.505 |
0.000 |
| z |
0.000 |
0.000 |
-1.862 |
|
| Polar |
|---|
| 3z2-r2 | -3.725 |
|---|
| x2-y2 | 7.249 |
|---|
| xy | 0.362 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
269.306 |
| (<r2>)1/2 |
16.411 |