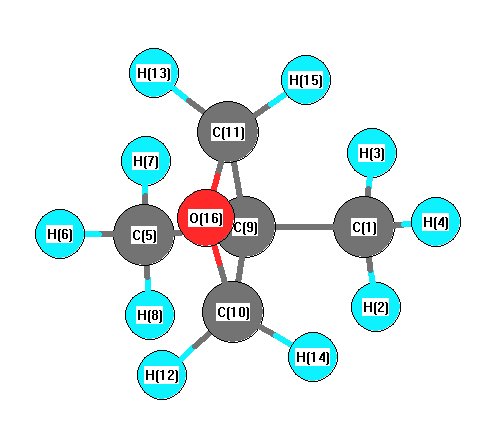
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3145 |
2993 |
27.34 |
|
|
|
| 2 |
A' |
3141 |
2989 |
29.98 |
|
|
|
| 3 |
A' |
3102 |
2953 |
67.40 |
|
|
|
| 4 |
A' |
3059 |
2911 |
23.15 |
|
|
|
| 5 |
A' |
3053 |
2906 |
34.35 |
|
|
|
| 6 |
A' |
3045 |
2898 |
20.42 |
|
|
|
| 7 |
A' |
1542 |
1467 |
3.54 |
|
|
|
| 8 |
A' |
1514 |
1441 |
4.75 |
|
|
|
| 9 |
A' |
1496 |
1424 |
4.68 |
|
|
|
| 10 |
A' |
1430 |
1361 |
5.07 |
|
|
|
| 11 |
A' |
1413 |
1345 |
12.34 |
|
|
|
| 12 |
A' |
1391 |
1324 |
0.39 |
|
|
|
| 13 |
A' |
1311 |
1248 |
0.53 |
|
|
|
| 14 |
A' |
1282 |
1220 |
8.93 |
|
|
|
| 15 |
A' |
1177 |
1120 |
1.23 |
|
|
|
| 16 |
A' |
1044 |
994 |
9.80 |
|
|
|
| 17 |
A' |
998 |
950 |
11.69 |
|
|
|
| 18 |
A' |
967 |
920 |
3.76 |
|
|
|
| 19 |
A' |
943 |
898 |
7.56 |
|
|
|
| 20 |
A' |
853 |
812 |
9.45 |
|
|
|
| 21 |
A' |
649 |
618 |
0.62 |
|
|
|
| 22 |
A' |
404 |
385 |
0.07 |
|
|
|
| 23 |
A' |
345 |
329 |
0.10 |
|
|
|
| 24 |
A' |
90 |
85 |
6.70 |
|
|
|
| 25 |
A" |
3135 |
2984 |
47.80 |
|
|
|
| 26 |
A" |
3131 |
2980 |
5.26 |
|
|
|
| 27 |
A" |
3102 |
2952 |
13.18 |
|
|
|
| 28 |
A" |
3036 |
2890 |
125.33 |
|
|
|
| 29 |
A" |
1526 |
1452 |
8.43 |
|
|
|
| 30 |
A" |
1509 |
1436 |
6.00 |
|
|
|
| 31 |
A" |
1495 |
1423 |
0.08 |
|
|
|
| 32 |
A" |
1314 |
1250 |
1.07 |
|
|
|
| 33 |
A" |
1235 |
1175 |
0.07 |
|
|
|
| 34 |
A" |
1166 |
1109 |
0.13 |
|
|
|
| 35 |
A" |
1072 |
1020 |
86.14 |
|
|
|
| 36 |
A" |
1062 |
1010 |
0.35 |
|
|
|
| 37 |
A" |
942 |
897 |
0.03 |
|
|
|
| 38 |
A" |
898 |
855 |
3.21 |
|
|
|
| 39 |
A" |
393 |
374 |
0.98 |
|
|
|
| 40 |
A" |
313 |
298 |
0.25 |
|
|
|
| 41 |
A" |
278 |
265 |
0.02 |
|
|
|
| 42 |
A" |
227 |
216 |
0.04 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31611.5 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 30087.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.627 |
|
|
|
| 2 |
H |
0.169 |
|
|
|
| 3 |
H |
0.169 |
|
|
|
| 4 |
H |
0.167 |
|
|
|
| 5 |
C |
-0.635 |
|
|
|
| 6 |
H |
0.176 |
|
|
|
| 7 |
H |
0.168 |
|
|
|
| 8 |
H |
0.168 |
|
|
|
| 9 |
C |
0.386 |
|
|
|
| 10 |
C |
-0.169 |
|
|
|
| 11 |
C |
-0.169 |
|
|
|
| 12 |
H |
0.149 |
|
|
|
| 13 |
H |
0.149 |
|
|
|
| 14 |
H |
0.143 |
|
|
|
| 15 |
H |
0.143 |
|
|
|
| 16 |
O |
-0.387 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.133 |
2.212 |
0.000 |
2.216 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.933 |
0.156 |
0.000 |
| y |
0.156 |
-44.334 |
0.000 |
| z |
0.000 |
0.000 |
-36.061 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.265 |
0.156 |
0.000 |
| y |
0.156 |
-7.337 |
0.000 |
| z |
0.000 |
0.000 |
5.073 |
|
| Polar |
|---|
| 3z2-r2 | 10.146 |
|---|
| x2-y2 | 6.401 |
|---|
| xy | 0.156 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
158.465 |
| (<r2>)1/2 |
12.588 |