Jump to
S1C2
Energy calculated at mPW1PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -342.328723 |
| Energy at 298.15K | |
| HF Energy | -342.328723 |
| Nuclear repulsion energy | 246.045237 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3094 |
2945 |
58.36 |
|
|
|
| 2 |
A1 |
1956 |
1862 |
681.04 |
|
|
|
| 3 |
A1 |
1561 |
1486 |
1.71 |
|
|
|
| 4 |
A1 |
1410 |
1342 |
0.36 |
|
|
|
| 5 |
A1 |
1240 |
1180 |
253.32 |
|
|
|
| 6 |
A1 |
966 |
919 |
9.16 |
|
|
|
| 7 |
A1 |
853 |
812 |
36.75 |
|
|
|
| 8 |
A1 |
729 |
694 |
0.83 |
|
|
|
| 9 |
A2 |
3126 |
2975 |
0.00 |
|
|
|
| 10 |
A2 |
1261 |
1200 |
0.00 |
|
|
|
| 11 |
A2 |
1176 |
1119 |
0.00 |
|
|
|
| 12 |
A2 |
179i |
171i |
0.00 |
|
|
|
| 13 |
B1 |
3147 |
2995 |
36.04 |
|
|
|
| 14 |
B1 |
1277 |
1216 |
2.09 |
|
|
|
| 15 |
B1 |
868 |
826 |
0.43 |
|
|
|
| 16 |
B1 |
765 |
728 |
24.88 |
|
|
|
| 17 |
B1 |
140 |
133 |
2.11 |
|
|
|
| 18 |
B2 |
3087 |
2938 |
22.69 |
|
|
|
| 19 |
B2 |
1547 |
1473 |
5.73 |
|
|
|
| 20 |
B2 |
1421 |
1352 |
15.74 |
|
|
|
| 21 |
B2 |
1225 |
1166 |
7.42 |
|
|
|
| 22 |
B2 |
1066 |
1015 |
333.62 |
|
|
|
| 23 |
B2 |
759 |
723 |
1.64 |
|
|
|
| 24 |
B2 |
508 |
483 |
0.04 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16500.8 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 15705.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31+G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.863 |
| O2 |
0.000 |
0.000 |
2.049 |
| O3 |
0.000 |
1.126 |
0.056 |
| O4 |
0.000 |
-1.126 |
0.056 |
| C5 |
0.000 |
0.774 |
-1.284 |
| C6 |
0.000 |
-0.774 |
-1.284 |
| H7 |
-0.888 |
1.188 |
-1.767 |
| H8 |
0.888 |
1.188 |
-1.767 |
| H9 |
0.888 |
-1.188 |
-1.767 |
| H10 |
-0.888 |
-1.188 |
-1.767 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1859 | 1.3855 | 1.3855 | 2.2821 | 2.2821 | 3.0202 | 3.0202 | 3.0202 | 3.0202 |
O2 | 1.1859 | | 2.2891 | 2.2891 | 3.4214 | 3.4214 | 4.0949 | 4.0949 | 4.0949 | 4.0949 | O3 | 1.3855 | 2.2891 | | 2.2523 | 1.3855 | 2.3247 | 2.0294 | 2.0294 | 3.0775 | 3.0775 | O4 | 1.3855 | 2.2891 | 2.2523 | | 2.3247 | 1.3855 | 3.0775 | 3.0775 | 2.0294 | 2.0294 | C5 | 2.2821 | 3.4214 | 1.3855 | 2.3247 | | 1.5472 | 1.0931 | 1.0931 | 2.2072 | 2.2072 | C6 | 2.2821 | 3.4214 | 2.3247 | 1.3855 | 1.5472 | | 2.2072 | 2.2072 | 1.0931 | 1.0931 | H7 | 3.0202 | 4.0949 | 2.0294 | 3.0775 | 1.0931 | 2.2072 | | 1.7762 | 2.9670 | 2.3765 | H8 | 3.0202 | 4.0949 | 2.0294 | 3.0775 | 1.0931 | 2.2072 | 1.7762 | | 2.3765 | 2.9670 | H9 | 3.0202 | 4.0949 | 3.0775 | 2.0294 | 2.2072 | 1.0931 | 2.9670 | 2.3765 | | 1.7762 | H10 | 3.0202 | 4.0949 | 3.0775 | 2.0294 | 2.2072 | 1.0931 | 2.3765 | 2.9670 | 1.7762 | |
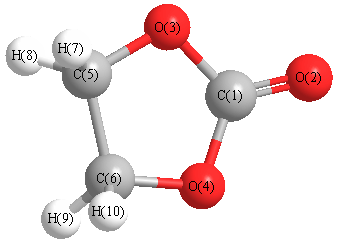 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.887 |
|
C1 |
O4 |
C6 |
110.887 |
| O2 |
C1 |
O3 |
125.628 |
|
O2 |
C1 |
O4 |
125.628 |
| O3 |
C1 |
O4 |
108.744 |
|
O3 |
C5 |
C6 |
104.741 |
| O3 |
C5 |
H7 |
109.364 |
|
O3 |
C5 |
H8 |
109.364 |
| O4 |
C6 |
C5 |
104.741 |
|
O4 |
C6 |
H9 |
109.364 |
| O4 |
C6 |
H10 |
109.364 |
|
C5 |
C6 |
H9 |
112.292 |
| C5 |
C6 |
H10 |
112.292 |
|
C6 |
C5 |
H7 |
112.292 |
| C6 |
C5 |
H8 |
112.292 |
|
H7 |
C5 |
H8 |
108.682 |
| H9 |
C6 |
H10 |
108.682 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.685 |
|
|
|
| 2 |
O |
-0.457 |
|
|
|
| 3 |
O |
-0.347 |
|
|
|
| 4 |
O |
-0.347 |
|
|
|
| 5 |
C |
-0.116 |
|
|
|
| 6 |
C |
-0.116 |
|
|
|
| 7 |
H |
0.175 |
|
|
|
| 8 |
H |
0.175 |
|
|
|
| 9 |
H |
0.175 |
|
|
|
| 10 |
H |
0.175 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.187 |
5.187 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.246 |
0.000 |
0.000 |
| y |
0.000 |
-36.629 |
0.000 |
| z |
0.000 |
0.000 |
-36.224 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.181 |
0.000 |
0.000 |
| y |
0.000 |
-2.394 |
0.000 |
| z |
0.000 |
0.000 |
-1.786 |
|
| Polar |
|---|
| 3z2-r2 | -3.573 |
|---|
| x2-y2 | 4.383 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.727 |
0.000 |
0.000 |
| y |
0.000 |
5.829 |
0.000 |
| z |
0.000 |
0.000 |
7.975 |
<r2> (average value of r
2) Å
2
| <r2> |
128.542 |
| (<r2>)1/2 |
11.338 |
Jump to
S1C1
Energy calculated at mPW1PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -342.329452 |
| Energy at 298.15K | -342.335848 |
| HF Energy | -342.329452 |
| Nuclear repulsion energy | 246.592117 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3152 |
3000 |
16.48 |
|
|
|
| 2 |
A |
3072 |
2924 |
32.51 |
|
|
|
| 3 |
A |
1960 |
1866 |
662.74 |
|
|
|
| 4 |
A |
1548 |
1474 |
1.94 |
|
|
|
| 5 |
A |
1414 |
1346 |
4.71 |
|
|
|
| 6 |
A |
1267 |
1206 |
30.21 |
|
|
|
| 7 |
A |
1234 |
1174 |
182.74 |
|
|
|
| 8 |
A |
1163 |
1107 |
32.55 |
|
|
|
| 9 |
A |
968 |
921 |
3.53 |
|
|
|
| 10 |
A |
848 |
807 |
40.32 |
|
|
|
| 11 |
A |
727 |
692 |
1.05 |
|
|
|
| 12 |
A |
133 |
126 |
0.57 |
|
|
|
| 13 |
B |
3162 |
3009 |
21.28 |
|
|
|
| 14 |
B |
3077 |
2928 |
42.34 |
|
|
|
| 15 |
B |
1539 |
1465 |
7.09 |
|
|
|
| 16 |
B |
1413 |
1345 |
13.74 |
|
|
|
| 17 |
B |
1281 |
1219 |
5.38 |
|
|
|
| 18 |
B |
1200 |
1143 |
5.80 |
|
|
|
| 19 |
B |
1060 |
1009 |
309.51 |
|
|
|
| 20 |
B |
908 |
865 |
8.40 |
|
|
|
| 21 |
B |
775 |
737 |
25.21 |
|
|
|
| 22 |
B |
696 |
662 |
1.02 |
|
|
|
| 23 |
B |
508 |
483 |
0.12 |
|
|
|
| 24 |
B |
149 |
142 |
1.84 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16626.6 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 15825.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31+G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.859 |
| O2 |
0.000 |
0.000 |
2.044 |
| O3 |
0.000 |
1.126 |
0.046 |
| O4 |
0.000 |
-1.126 |
0.046 |
| C5 |
0.191 |
0.742 |
-1.274 |
| C6 |
-0.191 |
-0.742 |
-1.274 |
| H7 |
-0.443 |
1.347 |
-1.924 |
| H8 |
1.240 |
0.896 |
-1.553 |
| H9 |
0.443 |
-1.347 |
-1.924 |
| H10 |
-1.240 |
-0.896 |
-1.553 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1854 | 1.3887 | 1.3887 | 2.2664 | 2.2664 | 3.1228 | 2.8559 | 3.1228 | 2.8559 |
O2 | 1.1854 | | 2.2932 | 2.2932 | 3.4056 | 3.4056 | 4.2135 | 3.9089 | 4.2135 | 3.9089 | O3 | 1.3887 | 2.2932 | | 2.2530 | 1.3887 | 2.2964 | 2.0313 | 2.0369 | 3.1933 | 2.8612 | O4 | 1.3887 | 2.2932 | 2.2530 | | 2.2964 | 1.3887 | 3.1933 | 2.8612 | 2.0313 | 2.0369 | C5 | 2.2664 | 3.4056 | 1.3887 | 2.2964 | | 1.5332 | 1.0908 | 1.0958 | 2.2027 | 2.1931 | C6 | 2.2664 | 3.4056 | 2.2964 | 1.3887 | 1.5332 | | 2.2027 | 2.1931 | 1.0908 | 1.0958 | H7 | 3.1228 | 4.2135 | 2.0313 | 3.1933 | 1.0908 | 2.2027 | | 1.7811 | 2.8366 | 2.4093 | H8 | 2.8559 | 3.9089 | 2.0369 | 2.8612 | 1.0958 | 2.1931 | 1.7811 | | 2.4093 | 3.0593 | H9 | 3.1228 | 4.2135 | 3.1933 | 2.0313 | 2.2027 | 1.0908 | 2.8366 | 2.4093 | | 1.7811 | H10 | 2.8559 | 3.9089 | 2.8612 | 2.0369 | 2.1931 | 1.0958 | 2.4093 | 3.0593 | 1.7811 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
109.378 |
|
C1 |
O4 |
C6 |
109.378 |
| O2 |
C1 |
O3 |
125.788 |
|
O2 |
C1 |
O4 |
125.788 |
| O3 |
C1 |
O4 |
108.424 |
|
O3 |
C5 |
C6 |
103.505 |
| O3 |
C5 |
H7 |
109.436 |
|
O3 |
C5 |
H8 |
109.574 |
| O4 |
C6 |
C5 |
103.505 |
|
O4 |
C6 |
H9 |
109.436 |
| O4 |
C6 |
H10 |
109.574 |
|
C5 |
C6 |
H9 |
113.085 |
| C5 |
C6 |
H10 |
111.989 |
|
C6 |
C5 |
H7 |
113.085 |
| C6 |
C5 |
H8 |
111.989 |
|
H7 |
C5 |
H8 |
109.085 |
| H9 |
C6 |
H10 |
109.085 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.696 |
|
|
|
| 2 |
O |
-0.466 |
|
|
|
| 3 |
O |
-0.346 |
|
|
|
| 4 |
O |
-0.346 |
|
|
|
| 5 |
C |
-0.123 |
|
|
|
| 6 |
C |
-0.123 |
|
|
|
| 7 |
H |
0.178 |
|
|
|
| 8 |
H |
0.176 |
|
|
|
| 9 |
H |
0.178 |
|
|
|
| 10 |
H |
0.176 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.131 |
5.131 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.328 |
0.294 |
0.000 |
| y |
0.294 |
-36.653 |
0.000 |
| z |
0.000 |
0.000 |
-36.012 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.005 |
0.294 |
0.000 |
| y |
0.294 |
-2.484 |
0.000 |
| z |
0.000 |
0.000 |
-1.521 |
|
| Polar |
|---|
| 3z2-r2 | -3.042 |
|---|
| x2-y2 | 4.326 |
|---|
| xy | 0.294 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.849 |
0.146 |
0.000 |
| y |
0.146 |
5.803 |
0.000 |
| z |
0.000 |
0.000 |
7.956 |
<r2> (average value of r
2) Å
2
| <r2> |
127.507 |
| (<r2>)1/2 |
11.292 |