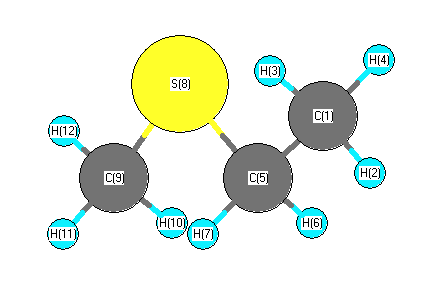
Vibrational Frequencies calculated at mPW1PW91/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3178 |
3045 |
4.68 |
|
|
|
| 2 |
A' |
3152 |
3021 |
17.13 |
|
|
|
| 3 |
A' |
3065 |
2937 |
21.16 |
|
|
|
| 4 |
A' |
3057 |
2930 |
12.12 |
|
|
|
| 5 |
A' |
3055 |
2928 |
52.05 |
|
|
|
| 6 |
A' |
1481 |
1420 |
2.53 |
|
|
|
| 7 |
A' |
1467 |
1406 |
0.66 |
|
|
|
| 8 |
A' |
1457 |
1396 |
11.32 |
|
|
|
| 9 |
A' |
1397 |
1339 |
3.48 |
|
|
|
| 10 |
A' |
1334 |
1279 |
2.14 |
|
|
|
| 11 |
A' |
1284 |
1231 |
31.69 |
|
|
|
| 12 |
A' |
1098 |
1052 |
2.21 |
|
|
|
| 13 |
A' |
1014 |
972 |
2.13 |
|
|
|
| 14 |
A' |
958 |
918 |
6.67 |
|
|
|
| 15 |
A' |
750 |
719 |
1.23 |
|
|
|
| 16 |
A' |
695 |
666 |
1.13 |
|
|
|
| 17 |
A' |
347 |
333 |
0.84 |
|
|
|
| 18 |
A' |
195 |
187 |
0.69 |
|
|
|
| 19 |
A" |
3158 |
3026 |
17.03 |
|
|
|
| 20 |
A" |
3150 |
3019 |
16.59 |
|
|
|
| 21 |
A" |
3109 |
2980 |
13.02 |
|
|
|
| 22 |
A" |
1469 |
1408 |
8.94 |
|
|
|
| 23 |
A" |
1443 |
1383 |
7.56 |
|
|
|
| 24 |
A" |
1257 |
1205 |
0.18 |
|
|
|
| 25 |
A" |
1038 |
995 |
0.11 |
|
|
|
| 26 |
A" |
960 |
920 |
4.04 |
|
|
|
| 27 |
A" |
790 |
757 |
3.95 |
|
|
|
| 28 |
A" |
248 |
238 |
0.15 |
|
|
|
| 29 |
A" |
168 |
161 |
0.43 |
|
|
|
| 30 |
A" |
75 |
72 |
0.53 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22924.8 cm
-1
Scaled (by 0.9583) Zero Point Vibrational Energy (zpe) 21968.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.080 |
|
|
|
| 2 |
H |
0.050 |
|
|
|
| 3 |
H |
0.059 |
|
|
|
| 4 |
H |
0.059 |
|
|
|
| 5 |
C |
-0.195 |
|
|
|
| 6 |
H |
0.069 |
|
|
|
| 7 |
H |
0.069 |
|
|
|
| 8 |
S |
-0.077 |
|
|
|
| 9 |
C |
-0.181 |
|
|
|
| 10 |
H |
0.074 |
|
|
|
| 11 |
H |
0.074 |
|
|
|
| 12 |
H |
0.079 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.941 |
-1.236 |
0.000 |
1.554 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
138.225 |
| (<r2>)1/2 |
11.757 |