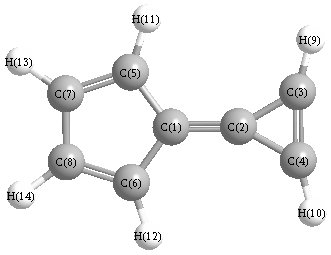
Vibrational Frequencies calculated at mPW1PW91/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3314 |
3179 |
13.77 |
|
|
|
| 2 |
A1 |
3252 |
3120 |
9.83 |
|
|
|
| 3 |
A1 |
3229 |
3098 |
7.32 |
|
|
|
| 4 |
A1 |
1836 |
1761 |
575.93 |
|
|
|
| 5 |
A1 |
1599 |
1533 |
126.02 |
|
|
|
| 6 |
A1 |
1507 |
1445 |
93.57 |
|
|
|
| 7 |
A1 |
1419 |
1361 |
114.29 |
|
|
|
| 8 |
A1 |
1236 |
1186 |
24.10 |
|
|
|
| 9 |
A1 |
1080 |
1036 |
13.60 |
|
|
|
| 10 |
A1 |
1035 |
993 |
10.14 |
|
|
|
| 11 |
A1 |
953 |
915 |
16.33 |
|
|
|
| 12 |
A1 |
893 |
857 |
37.81 |
|
|
|
| 13 |
A1 |
494 |
474 |
0.02 |
|
|
|
| 14 |
A2 |
941 |
902 |
0.00 |
|
|
|
| 15 |
A2 |
925 |
888 |
0.00 |
|
|
|
| 16 |
A2 |
711 |
682 |
0.00 |
|
|
|
| 17 |
A2 |
600 |
575 |
0.00 |
|
|
|
| 18 |
A2 |
213 |
204 |
0.00 |
|
|
|
| 19 |
B1 |
904 |
867 |
0.06 |
|
|
|
| 20 |
B1 |
767 |
736 |
62.87 |
|
|
|
| 21 |
B1 |
741 |
711 |
72.46 |
|
|
|
| 22 |
B1 |
637 |
611 |
9.83 |
|
|
|
| 23 |
B1 |
449 |
430 |
9.12 |
|
|
|
| 24 |
B1 |
137 |
132 |
6.58 |
|
|
|
| 25 |
B2 |
3273 |
3139 |
6.02 |
|
|
|
| 26 |
B2 |
3245 |
3113 |
20.75 |
|
|
|
| 27 |
B2 |
3219 |
3087 |
2.11 |
|
|
|
| 28 |
B2 |
1596 |
1531 |
0.70 |
|
|
|
| 29 |
B2 |
1357 |
1301 |
0.51 |
|
|
|
| 30 |
B2 |
1238 |
1187 |
2.60 |
|
|
|
| 31 |
B2 |
1135 |
1089 |
13.34 |
|
|
|
| 32 |
B2 |
1101 |
1056 |
9.89 |
|
|
|
| 33 |
B2 |
964 |
925 |
8.65 |
|
|
|
| 34 |
B2 |
838 |
804 |
1.54 |
|
|
|
| 35 |
B2 |
499 |
479 |
0.73 |
|
|
|
| 36 |
B2 |
148 |
142 |
1.90 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23741.1 cm
-1
Scaled (by 0.9592) Zero Point Vibrational Energy (zpe) 22772.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.081 |
|
|
|
| 2 |
C |
0.172 |
|
|
|
| 3 |
C |
-0.101 |
|
|
|
| 4 |
C |
-0.101 |
|
|
|
| 5 |
C |
-0.189 |
|
|
|
| 6 |
C |
-0.189 |
|
|
|
| 7 |
C |
-0.132 |
|
|
|
| 8 |
C |
-0.132 |
|
|
|
| 9 |
H |
0.124 |
|
|
|
| 10 |
H |
0.124 |
|
|
|
| 11 |
H |
0.082 |
|
|
|
| 12 |
H |
0.082 |
|
|
|
| 13 |
H |
0.090 |
|
|
|
| 14 |
H |
0.090 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
4.685 |
4.685 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-50.398 |
0.000 |
0.000 |
| y |
0.000 |
-41.132 |
0.000 |
| z |
0.000 |
0.000 |
-34.824 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-12.420 |
0.000 |
0.000 |
| y |
0.000 |
1.479 |
0.000 |
| z |
0.000 |
0.000 |
10.940 |
|
| Polar |
|---|
| 3z2-r2 | 21.880 |
|---|
| x2-y2 | -9.266 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.446 |
0.000 |
0.000 |
| y |
0.000 |
12.487 |
0.000 |
| z |
0.000 |
0.000 |
21.685 |
<r2> (average value of r
2) Å
2
| <r2> |
267.096 |
| (<r2>)1/2 |
16.343 |