Jump to
S1C2
Vibrational Frequencies calculated at mPW1PW91/3-21G*
Geometric Data calculated at mPW1PW91/3-21G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at mPW1PW91/3-21G*
| | hartrees |
|---|
| Energy at 0K | -621.857595 |
| Energy at 298.15K | -621.861483 |
| HF Energy | -621.857595 |
| Nuclear repulsion energy | 191.916725 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3456 |
3282 |
30.94 |
|
|
|
| 2 |
A' |
1261 |
1197 |
146.50 |
|
|
|
| 3 |
A' |
1115 |
1058 |
1.42 |
|
|
|
| 4 |
A' |
795 |
755 |
151.83 |
|
|
|
| 5 |
A' |
510 |
485 |
98.65 |
|
|
|
| 6 |
A' |
447 |
425 |
119.88 |
|
|
|
| 7 |
A' |
303 |
288 |
27.27 |
|
|
|
| 8 |
A" |
3454 |
3279 |
11.92 |
|
|
|
| 9 |
A" |
1071 |
1017 |
43.09 |
|
|
|
| 10 |
A" |
808 |
767 |
360.50 |
|
|
|
| 11 |
A" |
469 |
446 |
105.39 |
|
|
|
| 12 |
A" |
201 |
191 |
15.07 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6944.5 cm
-1
Scaled (by 0.9496) Zero Point Vibrational Energy (zpe) 6594.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/3-21G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.299 |
0.343 |
0.000 |
| O2 |
-1.038 |
0.958 |
0.000 |
| O3 |
0.299 |
-0.698 |
1.267 |
| O4 |
0.299 |
-0.698 |
-1.267 |
| H5 |
-0.629 |
-0.989 |
1.502 |
| H6 |
-0.629 |
-0.989 |
-1.502 |
Atom - Atom Distances (Å)
| |
S1 |
O2 |
O3 |
O4 |
H5 |
H6 |
| S1 | | 1.4716 | 1.6400 | 1.6400 | 2.2116 | 2.2116 |
O2 | 1.4716 | | 2.4769 | 2.4769 | 2.4926 | 2.4926 | O3 | 1.6400 | 2.4769 | | 2.5348 | 1.0003 | 2.9354 | O4 | 1.6400 | 2.4769 | 2.5348 | | 2.9354 | 1.0003 | H5 | 2.2116 | 2.4926 | 1.0003 | 2.9354 | | 3.0046 | H6 | 2.2116 | 2.4926 | 2.9354 | 1.0003 | 3.0046 | |
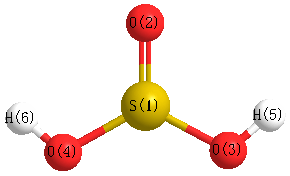 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
O3 |
H5 |
111.472 |
|
S1 |
O4 |
H6 |
111.472 |
| O2 |
S1 |
O3 |
105.373 |
|
O2 |
S1 |
O4 |
105.373 |
| O3 |
S1 |
O4 |
101.216 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
1.023 |
|
|
|
| 2 |
O |
-0.542 |
|
|
|
| 3 |
O |
-0.608 |
|
|
|
| 4 |
O |
-0.608 |
|
|
|
| 5 |
H |
0.368 |
|
|
|
| 6 |
H |
0.368 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.381 |
-1.613 |
0.000 |
2.124 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.951 |
6.088 |
0.000 |
| y |
6.088 |
-29.059 |
0.000 |
| z |
0.000 |
0.000 |
-28.002 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.421 |
6.088 |
0.000 |
| y |
6.088 |
-0.083 |
0.000 |
| z |
0.000 |
0.000 |
1.503 |
|
| Polar |
|---|
| 3z2-r2 | 3.007 |
|---|
| x2-y2 | -0.892 |
|---|
| xy | 6.088 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.216 |
-0.134 |
0.000 |
| y |
-0.134 |
3.165 |
0.000 |
| z |
0.000 |
0.000 |
3.647 |
<r2> (average value of r
2) Å
2
| <r2> |
79.569 |
| (<r2>)1/2 |
8.920 |