Jump to
S1C2
S1C3
S1C4
Energy calculated at MP2/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -8.553997 |
| Energy at 298.15K | |
| HF Energy | -8.438871 |
| Nuclear repulsion energy | 8.237703 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2272 |
2195 |
0.00 |
382.94 |
0.60 |
0.75 |
| 2 |
Σg |
683 |
660 |
0.00 |
38.79 |
0.33 |
0.49 |
| 3 |
Σu |
2266 |
2190 |
0.48 |
0.00 |
0.00 |
0.00 |
| 4 |
Πg |
601i |
580i |
0.00 |
92.11 |
0.75 |
0.86 |
| 4 |
Πg |
601i |
580i |
0.00 |
92.11 |
0.75 |
0.86 |
| 5 |
Πu |
379 |
367 |
10.42 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
379 |
367 |
10.42 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2389.1 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 2308.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.036 |
| Si2 |
0.000 |
0.000 |
-1.036 |
| H3 |
0.000 |
0.000 |
2.519 |
| H4 |
0.000 |
0.000 |
-2.519 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.0716 | 1.4830 | 3.5546 |
Si2 | 2.0716 | | 3.5546 | 1.4830 | H3 | 1.4830 | 3.5546 | | 5.0376 | H4 | 3.5546 | 1.4830 | 5.0376 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
S1C4
Energy calculated at MP2/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -8.586776 |
| Energy at 298.15K | |
| HF Energy | -8.472824 |
| Nuclear repulsion energy | 8.064858 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2102 |
2031 |
0.00 |
790.28 |
0.27 |
0.43 |
| 2 |
Ag |
614 |
593 |
0.00 |
2172.59 |
0.37 |
0.54 |
| 3 |
Ag |
576 |
557 |
0.00 |
3496.78 |
0.41 |
0.58 |
| 4 |
Au |
914 |
884 |
906.36 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2119 |
2048 |
267.69 |
0.00 |
0.00 |
0.00 |
| 6 |
Bu |
562 |
543 |
71.75 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3443.6 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 3327.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G
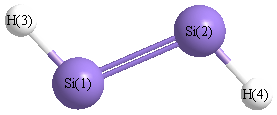
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.087 |
0.000 |
| Si2 |
0.000 |
-1.087 |
0.000 |
| H3 |
1.235 |
1.981 |
0.000 |
| H4 |
-1.235 |
-1.981 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1746 | 1.5240 | 3.3070 |
Si2 | 2.1746 | | 3.3070 | 1.5240 | H3 | 1.5240 | 3.3070 | | 4.6679 | H4 | 3.3070 | 1.5240 | 4.6679 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
125.884 |
|
Si2 |
Si1 |
H3 |
125.884 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S1C4
Energy calculated at MP2/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -8.590167 |
| Energy at 298.15K | |
| HF Energy | -8.491564 |
| Nuclear repulsion energy | 8.588376 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1509 |
1458 |
26.03 |
121.56 |
0.06 |
0.12 |
| 2 |
A1 |
905 |
874 |
108.25 |
37.32 |
0.24 |
0.38 |
| 3 |
A1 |
421 |
407 |
7.08 |
37.76 |
0.57 |
0.72 |
| 4 |
A2 |
970 |
937 |
0.00 |
5.03 |
0.75 |
0.86 |
| 5 |
B1 |
1396 |
1349 |
75.81 |
81.12 |
0.75 |
0.86 |
| 6 |
B2 |
1064 |
1028 |
544.11 |
1.60 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3132.4 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 3026.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.192 |
-0.053 |
| Si2 |
0.000 |
-1.192 |
-0.053 |
| H3 |
1.035 |
0.000 |
0.747 |
| H4 |
-1.035 |
0.000 |
0.747 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.3848 | 1.7704 | 1.7704 |
Si2 | 2.3848 | | 1.7704 | 1.7704 | H3 | 1.7704 | 1.7704 | | 2.0710 | H4 | 1.7704 | 1.7704 | 2.0710 | |
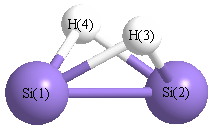 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
47.661 |
|
Si2 |
Si1 |
H3 |
47.661 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S1C3
Energy calculated at MP2/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -8.586422 |
| Energy at 298.15K | |
| HF Energy | -8.479840 |
| Nuclear repulsion energy | 8.303203 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2104 |
2033 |
150.32 |
396.29 |
0.54 |
0.70 |
| 2 |
A' |
1541 |
1489 |
120.10 |
185.05 |
0.34 |
0.51 |
| 3 |
A' |
922 |
891 |
282.36 |
6.82 |
0.71 |
0.83 |
| 4 |
A' |
557 |
538 |
35.09 |
12.32 |
0.70 |
0.83 |
| 5 |
A' |
422 |
408 |
24.79 |
0.94 |
0.58 |
0.74 |
| 6 |
A" |
240 |
232 |
59.07 |
4.06 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 2892.8 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 2795.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.059 |
-1.213 |
0.000 |
| Si2 |
0.059 |
1.030 |
0.000 |
| H3 |
-1.336 |
0.061 |
0.000 |
| H4 |
-0.329 |
2.498 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.2430 | 1.8893 | 3.7311 |
Si2 | 2.2430 | | 1.6990 | 1.5182 | H3 | 1.8893 | 1.6990 | | 2.6371 | H4 | 3.7311 | 1.5182 | 2.6371 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
165.194 |
|
Si2 |
Si1 |
H3 |
47.609 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability