Jump to
S1C2
Energy calculated at MP2/CEP-31G*
| | hartrees |
|---|
| Energy at 0K | -70.420786 |
| Energy at 298.15K | -70.425511 |
| HF Energy | -69.509769 |
| Nuclear repulsion energy | 123.705149 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3767 |
3577 |
92.68 |
|
|
|
| 2 |
A' |
3712 |
3524 |
85.65 |
|
|
|
| 3 |
A' |
3618 |
3435 |
89.46 |
|
|
|
| 4 |
A' |
1811 |
1719 |
110.04 |
|
|
|
| 5 |
A' |
1785 |
1695 |
473.81 |
|
|
|
| 6 |
A' |
1622 |
1540 |
92.51 |
|
|
|
| 7 |
A' |
1449 |
1376 |
14.78 |
|
|
|
| 8 |
A' |
1326 |
1259 |
45.99 |
|
|
|
| 9 |
A' |
1200 |
1140 |
332.17 |
|
|
|
| 10 |
A' |
1089 |
1034 |
4.72 |
|
|
|
| 11 |
A' |
785 |
745 |
6.00 |
|
|
|
| 12 |
A' |
598 |
568 |
67.89 |
|
|
|
| 13 |
A' |
524 |
497 |
0.17 |
|
|
|
| 14 |
A' |
412 |
391 |
4.41 |
|
|
|
| 15 |
A' |
264 |
251 |
13.45 |
|
|
|
| 16 |
A" |
813 |
772 |
7.81 |
|
|
|
| 17 |
A" |
682 |
647 |
148.95 |
|
|
|
| 18 |
A" |
630 |
598 |
5.09 |
|
|
|
| 19 |
A" |
425 |
404 |
0.17 |
|
|
|
| 20 |
A" |
205 |
194 |
297.51 |
|
|
|
| 21 |
A" |
27 |
26 |
4.38 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13372.0 cm
-1
Scaled (by 0.9494) Zero Point Vibrational Energy (zpe) 12695.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.762 |
0.000 |
| C2 |
-0.054 |
-0.794 |
0.000 |
| O3 |
-1.115 |
-1.433 |
0.000 |
| O4 |
1.053 |
1.409 |
0.000 |
| O5 |
-1.239 |
1.304 |
0.000 |
| N6 |
1.212 |
-1.325 |
0.000 |
| H7 |
1.314 |
-2.336 |
0.000 |
| H8 |
2.028 |
-0.717 |
0.000 |
| H9 |
-1.100 |
2.279 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5566 | 2.4614 | 1.2361 | 1.3521 | 2.4125 | 3.3652 | 2.5098 | 1.8736 |
C2 | 1.5566 | | 1.2389 | 2.4650 | 2.4096 | 1.3718 | 2.0613 | 2.0828 | 3.2459 | O3 | 2.4614 | 1.2389 | | 3.5741 | 2.7394 | 2.3292 | 2.5917 | 3.2233 | 3.7114 | O4 | 1.2361 | 2.4650 | 3.5741 | | 2.2943 | 2.7376 | 3.7541 | 2.3384 | 2.3222 | O5 | 1.3521 | 2.4096 | 2.7394 | 2.2943 | | 3.5934 | 4.4461 | 3.8412 | 0.9846 | N6 | 2.4125 | 1.3718 | 2.3292 | 2.7376 | 3.5934 | | 1.0171 | 1.0174 | 4.2808 | H7 | 3.3652 | 2.0613 | 2.5917 | 3.7541 | 4.4461 | 1.0171 | | 1.7698 | 5.2081 | H8 | 2.5098 | 2.0828 | 3.2233 | 2.3384 | 3.8412 | 1.0174 | 1.7698 | | 4.3308 | H9 | 1.8736 | 3.2459 | 3.7114 | 2.3222 | 0.9846 | 4.2808 | 5.2081 | 4.3308 | |
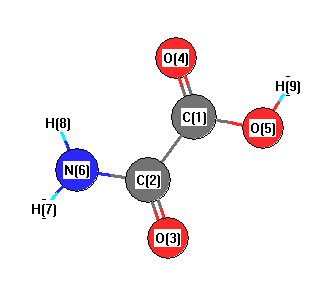 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
122.998 |
|
C1 |
C2 |
N6 |
110.779 |
| C1 |
O5 |
H9 |
105.535 |
|
C2 |
C1 |
O4 |
123.523 |
| C2 |
C1 |
O5 |
111.679 |
|
C2 |
N6 |
H7 |
118.518 |
| C2 |
N6 |
H8 |
120.598 |
|
O3 |
C2 |
N6 |
126.222 |
| O4 |
C1 |
O5 |
124.798 |
|
H7 |
N6 |
H8 |
120.883 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/CEP-31G*
| | hartrees |
|---|
| Energy at 0K | -70.424926 |
| Energy at 298.15K | -70.429977 |
| Nuclear repulsion energy | 124.539213 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3758 |
3568 |
102.85 |
|
|
|
| 2 |
A' |
3611 |
3428 |
157.87 |
|
|
|
| 3 |
A' |
3608 |
3425 |
77.07 |
|
|
|
| 4 |
A' |
1829 |
1737 |
194.17 |
|
|
|
| 5 |
A' |
1796 |
1705 |
340.04 |
|
|
|
| 6 |
A' |
1621 |
1539 |
56.32 |
|
|
|
| 7 |
A' |
1465 |
1391 |
177.57 |
|
|
|
| 8 |
A' |
1347 |
1279 |
411.43 |
|
|
|
| 9 |
A' |
1205 |
1144 |
13.95 |
|
|
|
| 10 |
A' |
1092 |
1037 |
3.94 |
|
|
|
| 11 |
A' |
803 |
763 |
12.75 |
|
|
|
| 12 |
A' |
614 |
582 |
11.09 |
|
|
|
| 13 |
A' |
536 |
509 |
2.48 |
|
|
|
| 14 |
A' |
405 |
384 |
6.10 |
|
|
|
| 15 |
A' |
271 |
257 |
38.85 |
|
|
|
| 16 |
A" |
806 |
765 |
0.01 |
|
|
|
| 17 |
A" |
715 |
679 |
133.96 |
|
|
|
| 18 |
A" |
653 |
620 |
5.34 |
|
|
|
| 19 |
A" |
439 |
417 |
34.28 |
|
|
|
| 20 |
A" |
297 |
282 |
273.50 |
|
|
|
| 21 |
A" |
100 |
95 |
0.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13485.1 cm
-1
Scaled (by 0.9494) Zero Point Vibrational Energy (zpe) 12802.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.005 |
-0.802 |
0.000 |
| C2 |
0.000 |
0.758 |
0.000 |
| O3 |
-1.085 |
1.377 |
0.000 |
| O4 |
1.035 |
-1.474 |
0.000 |
| O5 |
-1.244 |
-1.313 |
0.000 |
| N6 |
1.249 |
1.296 |
0.000 |
| H7 |
1.358 |
2.308 |
0.000 |
| H8 |
2.060 |
0.681 |
0.000 |
| H9 |
-1.849 |
-0.530 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5602 | 2.4365 | 1.2299 | 1.3495 | 2.4392 | 3.3914 | 2.5339 | 1.8742 |
C2 | 1.5602 | | 1.2487 | 2.4607 | 2.4158 | 1.3599 | 2.0605 | 2.0616 | 2.2535 | O3 | 2.4365 | 1.2487 | | 3.5530 | 2.6946 | 2.3348 | 2.6137 | 3.2208 | 2.0546 | O4 | 1.2299 | 2.4607 | 3.5530 | | 2.2849 | 2.7786 | 3.7958 | 2.3860 | 3.0350 | O5 | 1.3495 | 2.4158 | 2.6946 | 2.2849 | | 3.6084 | 4.4585 | 3.8587 | 0.9895 | N6 | 2.4392 | 1.3599 | 2.3348 | 2.7786 | 3.6084 | | 1.0175 | 1.0184 | 3.5961 | H7 | 3.3914 | 2.0605 | 2.6137 | 3.7958 | 4.4585 | 1.0175 | | 1.7724 | 4.2823 | H8 | 2.5339 | 2.0616 | 3.2208 | 2.3860 | 3.8587 | 1.0184 | 1.7724 | | 4.0923 | H9 | 1.8742 | 2.2535 | 2.0546 | 3.0350 | 0.9895 | 3.5961 | 4.2823 | 4.0923 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.911 |
|
C1 |
C2 |
N6 |
113.115 |
| C1 |
O5 |
H9 |
105.470 |
|
C2 |
C1 |
O4 |
123.317 |
| C2 |
C1 |
O5 |
112.042 |
|
C2 |
N6 |
H7 |
119.457 |
| C2 |
N6 |
H8 |
119.493 |
|
O3 |
C2 |
N6 |
126.974 |
| O4 |
C1 |
O5 |
124.640 |
|
H7 |
N6 |
H8 |
121.050 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability