Jump to
S1C2
Energy calculated at MP2/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -25.687593 |
| Energy at 298.15K | -25.692725 |
| HF Energy | -25.337256 |
| Nuclear repulsion energy | 55.113440 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3198 |
3121 |
0.00 |
|
|
|
| 2 |
Ag |
3097 |
3022 |
0.00 |
|
|
|
| 3 |
Ag |
3081 |
3007 |
0.00 |
|
|
|
| 4 |
Ag |
1655 |
1615 |
0.00 |
|
|
|
| 5 |
Ag |
1494 |
1458 |
0.00 |
|
|
|
| 6 |
Ag |
1317 |
1286 |
0.00 |
|
|
|
| 7 |
Ag |
1232 |
1202 |
0.00 |
|
|
|
| 8 |
Ag |
887 |
866 |
0.00 |
|
|
|
| 9 |
Ag |
508 |
496 |
0.00 |
|
|
|
| 10 |
Au |
982 |
959 |
68.25 |
|
|
|
| 11 |
Au |
811 |
792 |
91.06 |
|
|
|
| 12 |
Au |
479 |
468 |
12.88 |
|
|
|
| 13 |
Au |
133 |
129 |
0.04 |
|
|
|
| 14 |
Bg |
935 |
913 |
0.00 |
|
|
|
| 15 |
Bg |
814 |
794 |
0.00 |
|
|
|
| 16 |
Bg |
669 |
653 |
0.00 |
|
|
|
| 17 |
Bu |
3198 |
3122 |
60.62 |
|
|
|
| 18 |
Bu |
3101 |
3026 |
21.59 |
|
|
|
| 19 |
Bu |
3084 |
3010 |
37.89 |
|
|
|
| 20 |
Bu |
1597 |
1558 |
12.90 |
|
|
|
| 21 |
Bu |
1414 |
1380 |
1.20 |
|
|
|
| 22 |
Bu |
1330 |
1298 |
2.10 |
|
|
|
| 23 |
Bu |
1011 |
987 |
3.32 |
|
|
|
| 24 |
Bu |
291 |
284 |
1.38 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18159.1 cm
-1
Scaled (by 0.976) Zero Point Vibrational Energy (zpe) 17723.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-121G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.746 |
0.000 |
| C2 |
0.000 |
-0.746 |
0.000 |
| C3 |
1.145 |
1.507 |
0.000 |
| C4 |
-1.145 |
-1.507 |
0.000 |
| H5 |
-0.981 |
1.241 |
0.000 |
| H6 |
0.981 |
-1.241 |
0.000 |
| H7 |
1.106 |
2.599 |
0.000 |
| H8 |
2.138 |
1.045 |
0.000 |
| H9 |
-1.106 |
-2.599 |
0.000 |
| H10 |
-2.138 |
-1.045 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4929 | 1.3749 | 2.5280 | 1.0984 | 2.2160 | 2.1577 | 2.1588 | 3.5236 | 2.7891 |
C2 | 1.4929 | | 2.5280 | 1.3749 | 2.2160 | 1.0984 | 3.5236 | 2.7891 | 2.1577 | 2.1588 | C3 | 1.3749 | 2.5280 | | 3.7859 | 2.1427 | 2.7529 | 1.0926 | 1.0955 | 4.6830 | 4.1584 | C4 | 2.5280 | 1.3749 | 3.7859 | | 2.7529 | 2.1427 | 4.6830 | 4.1584 | 1.0926 | 1.0955 | H5 | 1.0984 | 2.2160 | 2.1427 | 2.7529 | | 3.1631 | 2.4900 | 3.1251 | 3.8418 | 2.5615 | H6 | 2.2160 | 1.0984 | 2.7529 | 2.1427 | 3.1631 | | 3.8418 | 2.5615 | 2.4900 | 3.1251 | H7 | 2.1577 | 3.5236 | 1.0926 | 4.6830 | 2.4900 | 3.8418 | | 1.8660 | 5.6493 | 4.8786 | H8 | 2.1588 | 2.7891 | 1.0955 | 4.1584 | 3.1251 | 2.5615 | 1.8660 | | 4.8786 | 4.7592 | H9 | 3.5236 | 2.1577 | 4.6830 | 1.0926 | 3.8418 | 2.4900 | 5.6493 | 4.8786 | | 1.8660 | H10 | 2.7891 | 2.1588 | 4.1584 | 1.0955 | 2.5615 | 3.1251 | 4.8786 | 4.7592 | 1.8660 | |
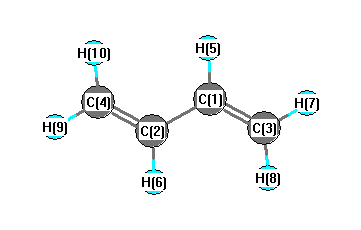 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
123.603 |
|
C1 |
C2 |
H6 |
116.740 |
| C1 |
C3 |
H7 |
121.543 |
|
C1 |
C3 |
H8 |
121.412 |
| C2 |
C1 |
C3 |
123.603 |
|
C2 |
C1 |
H5 |
116.740 |
| C2 |
C4 |
H9 |
121.543 |
|
C2 |
C4 |
H10 |
121.412 |
| C3 |
C1 |
H5 |
119.657 |
|
C4 |
C2 |
H6 |
119.657 |
| H7 |
C3 |
H8 |
117.044 |
|
H9 |
C4 |
H10 |
117.044 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -25.684426 |
| Energy at 298.15K | -25.689740 |
| HF Energy | -25.331813 |
| Nuclear repulsion energy | 55.736449 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3202 |
3125 |
12.52 |
|
|
|
| 2 |
A |
3114 |
3039 |
16.16 |
|
|
|
| 3 |
A |
3087 |
3013 |
24.35 |
|
|
|
| 4 |
A |
1624 |
1585 |
2.33 |
|
|
|
| 5 |
A |
1482 |
1446 |
5.82 |
|
|
|
| 6 |
A |
1333 |
1301 |
0.06 |
|
|
|
| 7 |
A |
1074 |
1048 |
0.50 |
|
|
|
| 8 |
A |
973 |
949 |
8.16 |
|
|
|
| 9 |
A |
879 |
858 |
0.00 |
|
|
|
| 10 |
A |
857 |
836 |
14.65 |
|
|
|
| 11 |
A |
727 |
710 |
3.46 |
|
|
|
| 12 |
A |
273 |
267 |
0.01 |
|
|
|
| 13 |
A |
189 |
184 |
0.04 |
|
|
|
| 14 |
B |
3199 |
3122 |
47.21 |
|
|
|
| 15 |
B |
3100 |
3026 |
21.85 |
|
|
|
| 16 |
B |
3084 |
3010 |
5.28 |
|
|
|
| 17 |
B |
1620 |
1581 |
4.48 |
|
|
|
| 18 |
B |
1449 |
1415 |
0.27 |
|
|
|
| 19 |
B |
1305 |
1274 |
0.02 |
|
|
|
| 20 |
B |
1100 |
1073 |
4.95 |
|
|
|
| 21 |
B |
995 |
971 |
52.38 |
|
|
|
| 22 |
B |
853 |
832 |
85.54 |
|
|
|
| 23 |
B |
627 |
612 |
9.32 |
|
|
|
| 24 |
B |
457 |
446 |
11.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18300.5 cm
-1
Scaled (by 0.976) Zero Point Vibrational Energy (zpe) 17861.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-121G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.136 |
0.739 |
0.566 |
| C2 |
-0.136 |
-0.739 |
0.566 |
| C3 |
-0.136 |
1.564 |
-0.500 |
| C4 |
0.136 |
-1.564 |
-0.500 |
| H5 |
0.562 |
1.167 |
1.482 |
| H6 |
-0.562 |
-1.167 |
1.482 |
| H7 |
0.097 |
2.632 |
-0.467 |
| H8 |
-0.610 |
1.181 |
-1.409 |
| H9 |
-0.097 |
-2.632 |
-0.467 |
| H10 |
0.610 |
-1.181 |
-1.409 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.5036 | 1.3748 | 2.5383 | 1.0973 | 2.2272 | 2.1560 | 2.1565 | 3.5334 | 2.7947 |
C2 | 1.5036 | | 2.5383 | 1.3748 | 2.2272 | 1.0973 | 3.5334 | 2.7947 | 2.1560 | 2.1565 | C3 | 1.3748 | 2.5383 | | 3.1407 | 2.1389 | 3.4014 | 1.0930 | 1.0946 | 4.1965 | 2.9864 | C4 | 2.5383 | 1.3748 | 3.1407 | | 3.4014 | 2.1389 | 4.1965 | 2.9864 | 1.0930 | 1.0946 | H5 | 1.0973 | 2.2272 | 2.1389 | 3.4014 | | 2.5896 | 2.4824 | 3.1198 | 4.3199 | 3.7246 | H6 | 2.2272 | 1.0973 | 3.4014 | 2.1389 | 2.5896 | | 4.3199 | 3.7246 | 2.4824 | 3.1198 | H7 | 2.1560 | 3.5334 | 1.0930 | 4.1965 | 2.4824 | 4.3199 | | 1.8690 | 5.2670 | 3.9603 | H8 | 2.1565 | 2.7947 | 1.0946 | 2.9864 | 3.1198 | 3.7246 | 1.8690 | | 3.9603 | 2.6575 | H9 | 3.5334 | 2.1560 | 4.1965 | 1.0930 | 4.3199 | 2.4824 | 5.2670 | 3.9603 | | 1.8690 | H10 | 2.7947 | 2.1565 | 2.9864 | 1.0946 | 3.7246 | 3.1198 | 3.9603 | 2.6575 | 1.8690 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
123.667 |
|
C1 |
C2 |
H6 |
116.944 |
| C1 |
C3 |
H7 |
121.356 |
|
C1 |
C3 |
H8 |
121.261 |
| C2 |
C1 |
C3 |
123.667 |
|
C2 |
C1 |
H5 |
116.944 |
| C2 |
C4 |
H9 |
121.356 |
|
C2 |
C4 |
H10 |
121.261 |
| C3 |
C1 |
H5 |
119.383 |
|
C4 |
C2 |
H6 |
119.383 |
| H7 |
C3 |
H8 |
117.376 |
|
H9 |
C4 |
H10 |
117.376 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability