Jump to
S1C2
Energy calculated at MP2/SDD
| | hartrees |
|---|
| Energy at 0K | -357.085749 |
| Energy at 298.15K | -357.090823 |
| HF Energy | -356.425539 |
| Nuclear repulsion energy | 225.546643 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3745 |
3615 |
72.60 |
|
|
|
| 2 |
A' |
3603 |
3478 |
74.78 |
|
|
|
| 3 |
A' |
3588 |
3464 |
86.08 |
|
|
|
| 4 |
A' |
1696 |
1638 |
194.08 |
|
|
|
| 5 |
A' |
1631 |
1574 |
135.89 |
|
|
|
| 6 |
A' |
1604 |
1548 |
192.21 |
|
|
|
| 7 |
A' |
1392 |
1344 |
1.26 |
|
|
|
| 8 |
A' |
1304 |
1259 |
80.87 |
|
|
|
| 9 |
A' |
1110 |
1072 |
109.94 |
|
|
|
| 10 |
A' |
1077 |
1040 |
227.91 |
|
|
|
| 11 |
A' |
737 |
712 |
0.82 |
|
|
|
| 12 |
A' |
574 |
554 |
69.86 |
|
|
|
| 13 |
A' |
522 |
504 |
1.53 |
|
|
|
| 14 |
A' |
403 |
389 |
8.57 |
|
|
|
| 15 |
A' |
262 |
253 |
11.86 |
|
|
|
| 16 |
A" |
782 |
755 |
0.68 |
|
|
|
| 17 |
A" |
648 |
625 |
391.57 |
|
|
|
| 18 |
A" |
608 |
587 |
13.58 |
|
|
|
| 19 |
A" |
599 |
578 |
206.35 |
|
|
|
| 20 |
A" |
407 |
393 |
13.11 |
|
|
|
| 21 |
A" |
65 |
63 |
7.59 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13178.3 cm
-1
Scaled (by 0.9653) Zero Point Vibrational Energy (zpe) 12721.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/SDD
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.757 |
0.000 |
| C2 |
-0.046 |
-0.795 |
0.000 |
| O3 |
-1.121 |
-1.467 |
0.000 |
| O4 |
1.071 |
1.427 |
0.000 |
| O5 |
-1.266 |
1.322 |
0.000 |
| N6 |
1.233 |
-1.327 |
0.000 |
| H7 |
1.349 |
-2.336 |
0.000 |
| H8 |
2.047 |
-0.718 |
0.000 |
| H9 |
-1.218 |
2.312 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5532 | 2.4912 | 1.2634 | 1.3867 | 2.4220 | 3.3746 | 2.5235 | 1.9747 |
C2 | 1.5532 | | 1.2678 | 2.4875 | 2.4439 | 1.3858 | 2.0786 | 2.0949 | 3.3203 | O3 | 2.4912 | 1.2678 | | 3.6311 | 2.7934 | 2.3588 | 2.6184 | 3.2559 | 3.7800 | O4 | 1.2634 | 2.4875 | 3.6311 | | 2.3399 | 2.7591 | 3.7735 | 2.3570 | 2.4542 | O5 | 1.3867 | 2.4439 | 2.7934 | 2.3399 | | 3.6426 | 4.4970 | 3.8915 | 0.9903 | N6 | 2.4220 | 1.3858 | 2.3588 | 2.7591 | 3.6426 | | 1.0155 | 1.0165 | 4.3874 | H7 | 3.3746 | 2.0786 | 2.6184 | 3.7735 | 4.4970 | 1.0155 | | 1.7622 | 5.3093 | H8 | 2.5235 | 2.0949 | 3.2559 | 2.3570 | 3.8915 | 1.0165 | 1.7622 | | 4.4543 | H9 | 1.9747 | 3.3203 | 3.7800 | 2.4542 | 0.9903 | 4.3874 | 5.3093 | 4.4543 | |
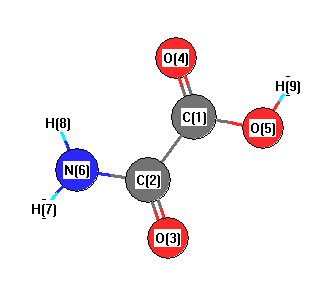 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
123.723 |
|
C1 |
C2 |
N6 |
110.863 |
| C1 |
O5 |
H9 |
111.255 |
|
C2 |
C1 |
O4 |
123.730 |
| C2 |
C1 |
O5 |
112.338 |
|
C2 |
N6 |
H7 |
119.098 |
| C2 |
N6 |
H8 |
120.624 |
|
O3 |
C2 |
N6 |
125.414 |
| O4 |
C1 |
O5 |
123.931 |
|
H7 |
N6 |
H8 |
120.279 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/SDD
| | hartrees |
|---|
| Energy at 0K | -357.089997 |
| Energy at 298.15K | -357.095241 |
| Nuclear repulsion energy | 226.580065 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3735 |
3606 |
79.22 |
|
|
|
| 2 |
A' |
3580 |
3456 |
106.31 |
|
|
|
| 3 |
A' |
3541 |
3418 |
104.33 |
|
|
|
| 4 |
A' |
1704 |
1645 |
220.83 |
|
|
|
| 5 |
A' |
1668 |
1611 |
230.24 |
|
|
|
| 6 |
A' |
1623 |
1566 |
1.58 |
|
|
|
| 7 |
A' |
1394 |
1345 |
18.31 |
|
|
|
| 8 |
A' |
1270 |
1226 |
512.72 |
|
|
|
| 9 |
A' |
1139 |
1099 |
52.05 |
|
|
|
| 10 |
A' |
1098 |
1060 |
9.43 |
|
|
|
| 11 |
A' |
749 |
723 |
19.71 |
|
|
|
| 12 |
A' |
594 |
573 |
10.87 |
|
|
|
| 13 |
A' |
532 |
513 |
2.07 |
|
|
|
| 14 |
A' |
393 |
380 |
4.44 |
|
|
|
| 15 |
A' |
261 |
252 |
39.55 |
|
|
|
| 16 |
A" |
778 |
751 |
1.56 |
|
|
|
| 17 |
A" |
682 |
658 |
321.49 |
|
|
|
| 18 |
A" |
651 |
628 |
261.30 |
|
|
|
| 19 |
A" |
633 |
611 |
20.08 |
|
|
|
| 20 |
A" |
421 |
406 |
4.58 |
|
|
|
| 21 |
A" |
110 |
106 |
10.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13276.5 cm
-1
Scaled (by 0.9653) Zero Point Vibrational Energy (zpe) 12815.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/SDD
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.019 |
-0.797 |
0.000 |
| C2 |
0.000 |
0.764 |
0.000 |
| O3 |
-1.114 |
1.389 |
0.000 |
| O4 |
1.072 |
-1.479 |
0.000 |
| O5 |
-1.253 |
-1.347 |
0.000 |
| N6 |
1.252 |
1.324 |
0.000 |
| H7 |
1.356 |
2.335 |
0.000 |
| H8 |
2.074 |
0.724 |
0.000 |
| H9 |
-1.945 |
-0.633 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5608 | 2.4621 | 1.2546 | 1.3860 | 2.4535 | 3.4049 | 2.5564 | 1.9702 |
C2 | 1.5608 | | 1.2775 | 2.4855 | 2.4548 | 1.3718 | 2.0749 | 2.0742 | 2.3944 | O3 | 2.4621 | 1.2775 | | 3.6059 | 2.7395 | 2.3671 | 2.6445 | 3.2565 | 2.1862 | O4 | 1.2546 | 2.4855 | 3.6059 | | 2.3290 | 2.8085 | 3.8239 | 2.4196 | 3.1328 | O5 | 1.3860 | 2.4548 | 2.7395 | 2.3290 | | 3.6622 | 4.5122 | 3.9188 | 0.9936 | N6 | 2.4535 | 1.3718 | 2.3671 | 2.8085 | 3.6622 | | 1.0158 | 1.0176 | 3.7484 | H7 | 3.4049 | 2.0749 | 2.6445 | 3.8239 | 4.5122 | 1.0158 | | 1.7637 | 4.4384 | H8 | 2.5564 | 2.0742 | 3.2565 | 2.4196 | 3.9188 | 1.0176 | 1.7637 | | 4.2413 | H9 | 1.9702 | 2.3944 | 2.1862 | 3.1328 | 0.9936 | 3.7484 | 4.4384 | 4.2413 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.994 |
|
C1 |
C2 |
N6 |
113.416 |
| C1 |
O5 |
H9 |
110.706 |
|
C2 |
C1 |
O4 |
123.608 |
| C2 |
C1 |
O5 |
112.691 |
|
C2 |
N6 |
H7 |
119.950 |
| C2 |
N6 |
H8 |
119.740 |
|
O3 |
C2 |
N6 |
126.591 |
| O4 |
C1 |
O5 |
123.701 |
|
H7 |
N6 |
H8 |
120.310 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability