Jump to
S1C2
Energy calculated at MP2/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -274.887988 |
| Energy at 298.15K | |
| HF Energy | -274.158258 |
| Nuclear repulsion energy | 116.337413 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2018 |
1935 |
50.11 |
85.49 |
0.09 |
0.16 |
| 2 |
A1 |
743 |
712 |
15.66 |
15.38 |
0.08 |
0.14 |
| 3 |
A1 |
546 |
524 |
61.44 |
1.52 |
0.12 |
0.21 |
| 4 |
A1 |
106 |
102 |
12.07 |
1.85 |
0.62 |
0.77 |
| 5 |
A2 |
476 |
457 |
0.00 |
2.55 |
0.75 |
0.86 |
| 6 |
B1 |
494 |
473 |
95.24 |
0.23 |
0.75 |
0.86 |
| 7 |
B2 |
2024 |
1941 |
1012.88 |
4.32 |
0.75 |
0.86 |
| 8 |
B2 |
1159 |
1111 |
100.53 |
0.92 |
0.75 |
0.86 |
| 9 |
B2 |
462 |
443 |
14.03 |
1.39 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4013.9 cm
-1
Scaled (by 0.959) Zero Point Vibrational Energy (zpe) 3849.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/aug-cc-pVDZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.614 |
| B2 |
0.000 |
1.235 |
0.076 |
| B3 |
0.000 |
-1.235 |
0.076 |
| O4 |
0.000 |
2.384 |
-0.355 |
| O5 |
0.000 |
-2.384 |
-0.355 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3471 | 1.3471 | 2.5730 | 2.5730 |
B2 | 1.3471 | | 2.4701 | 1.2268 | 3.6443 | B3 | 1.3471 | 2.4701 | | 3.6443 | 1.2268 | O4 | 2.5730 | 1.2268 | 3.6443 | | 4.7675 | O5 | 2.5730 | 3.6443 | 1.2268 | 4.7675 | |
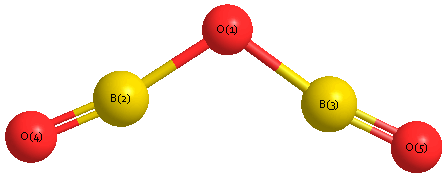 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.014 |
|
O1 |
B3 |
O5 |
177.014 |
| B2 |
O1 |
B3 |
132.933 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -274.884040 |
| Energy at 298.15K | |
| HF Energy | -274.156254 |
| Nuclear repulsion energy | 115.426121 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2025 |
1942 |
0.00 |
120.17 |
0.12 |
0.21 |
| 2 |
Σg |
647 |
620 |
0.00 |
15.44 |
0.11 |
0.20 |
| 3 |
Σu |
2063 |
1978 |
1569.04 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1253 |
1201 |
113.70 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
483 |
463 |
0.00 |
2.80 |
0.75 |
0.86 |
| 5 |
Πg |
483 |
463 |
0.00 |
2.80 |
0.75 |
0.86 |
| 6 |
Πu |
448 |
430 |
107.02 |
0.00 |
0.00 |
0.00 |
| 6 |
Πu |
448 |
430 |
107.02 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
112i |
107i |
3.01 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
112i |
107i |
3.01 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3813.0 cm
-1
Scaled (by 0.959) Zero Point Vibrational Energy (zpe) 3656.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/aug-cc-pVDZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.326 |
| B3 |
0.000 |
0.000 |
-1.326 |
| O4 |
0.000 |
0.000 |
2.555 |
| O5 |
0.000 |
0.000 |
-2.555 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3265 | 1.3265 | 2.5545 | 2.5545 |
B2 | 1.3265 | | 2.6529 | 1.2281 | 3.8810 | B3 | 1.3265 | 2.6529 | | 3.8810 | 1.2281 | O4 | 2.5545 | 1.2281 | 3.8810 | | 5.1091 | O5 | 2.5545 | 3.8810 | 1.2281 | 5.1091 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability