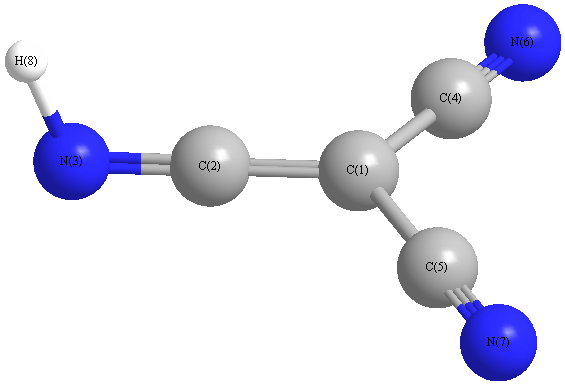
Vibrational Frequencies calculated at MP2/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3602 |
3402 |
168.48 |
377.16 |
0.31 |
0.47 |
| 2 |
A' |
2216 |
2093 |
49.25 |
205.25 |
0.11 |
0.19 |
| 3 |
A' |
2167 |
2046 |
415.62 |
5.68 |
0.49 |
0.66 |
| 4 |
A' |
1337 |
1263 |
2.32 |
51.95 |
0.24 |
0.39 |
| 5 |
A' |
803 |
758 |
540.80 |
16.74 |
0.53 |
0.69 |
| 6 |
A' |
654 |
618 |
1.08 |
13.98 |
0.09 |
0.17 |
| 7 |
A' |
595 |
562 |
17.82 |
4.63 |
0.60 |
0.75 |
| 8 |
A' |
579 |
547 |
0.24 |
0.34 |
0.51 |
0.68 |
| 9 |
A' |
447 |
423 |
41.29 |
2.49 |
0.39 |
0.56 |
| 10 |
A' |
172 |
163 |
6.89 |
0.46 |
0.73 |
0.84 |
| 11 |
A' |
132 |
125 |
8.73 |
7.09 |
0.73 |
0.84 |
| 12 |
A" |
2221 |
2097 |
2.72 |
144.62 |
0.75 |
0.86 |
| 13 |
A" |
1226 |
1158 |
0.33 |
8.30 |
0.75 |
0.86 |
| 14 |
A" |
748 |
707 |
85.67 |
1.30 |
0.75 |
0.86 |
| 15 |
A" |
625 |
590 |
2.40 |
0.32 |
0.75 |
0.86 |
| 16 |
A" |
406 |
383 |
6.18 |
3.43 |
0.75 |
0.86 |
| 17 |
A" |
387 |
365 |
0.32 |
0.16 |
0.75 |
0.86 |
| 18 |
A" |
128 |
121 |
0.02 |
9.13 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 9221.6 cm
-1
Scaled (by 0.9445) Zero Point Vibrational Energy (zpe) 8709.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.