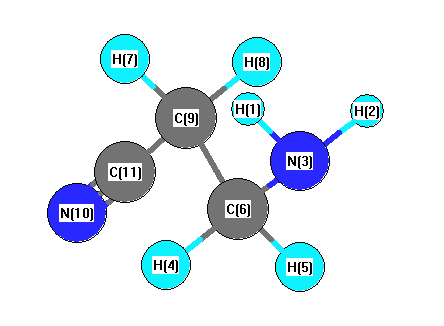
Vibrational Frequencies calculated at MP2/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3545 |
3360 |
1.46 |
|
|
|
| 2 |
A' |
3117 |
2954 |
24.32 |
|
|
|
| 3 |
A' |
3101 |
2940 |
2.53 |
|
|
|
| 4 |
A' |
2212 |
2097 |
0.04 |
|
|
|
| 5 |
A' |
1666 |
1579 |
29.54 |
|
|
|
| 6 |
A' |
1514 |
1435 |
4.47 |
|
|
|
| 7 |
A' |
1492 |
1415 |
3.08 |
|
|
|
| 8 |
A' |
1426 |
1352 |
15.14 |
|
|
|
| 9 |
A' |
1319 |
1250 |
1.75 |
|
|
|
| 10 |
A' |
1135 |
1076 |
12.28 |
|
|
|
| 11 |
A' |
1047 |
992 |
36.45 |
|
|
|
| 12 |
A' |
961 |
911 |
6.19 |
|
|
|
| 13 |
A' |
849 |
805 |
196.29 |
|
|
|
| 14 |
A' |
515 |
488 |
1.47 |
|
|
|
| 15 |
A' |
376 |
356 |
7.91 |
|
|
|
| 16 |
A' |
156 |
147 |
6.77 |
|
|
|
| 17 |
A" |
3647 |
3457 |
2.63 |
|
|
|
| 18 |
A" |
3178 |
3013 |
18.67 |
|
|
|
| 19 |
A" |
3152 |
2988 |
0.00 |
|
|
|
| 20 |
A" |
1411 |
1337 |
0.12 |
|
|
|
| 21 |
A" |
1324 |
1255 |
0.14 |
|
|
|
| 22 |
A" |
1169 |
1108 |
0.05 |
|
|
|
| 23 |
A" |
991 |
939 |
0.49 |
|
|
|
| 24 |
A" |
777 |
736 |
0.45 |
|
|
|
| 25 |
A" |
362 |
343 |
0.14 |
|
|
|
| 26 |
A" |
303 |
287 |
48.93 |
|
|
|
| 27 |
A" |
106 |
100 |
1.53 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20423.4 cm
-1
Scaled (by 0.9479) Zero Point Vibrational Energy (zpe) 19359.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.