Jump to
S1C2
S1C3
S1C4
Energy calculated at MP2/TZVP
| | hartrees |
|---|
| Energy at 0K | -579.058613 |
| Energy at 298.15K | |
| HF Energy | -578.862834 |
| Nuclear repulsion energy | 66.649240 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2416 |
2290 |
0.00 |
311.25 |
0.31 |
0.47 |
| 2 |
Σg |
734 |
696 |
0.00 |
53.49 |
0.23 |
0.37 |
| 3 |
Σu |
2413 |
2288 |
0.65 |
0.00 |
0.00 |
0.00 |
| 4 |
Πg |
596i |
565i |
0.00 |
31.91 |
0.75 |
0.86 |
| 4 |
Πg |
596i |
565i |
0.00 |
31.91 |
0.75 |
0.86 |
| 5 |
Πu |
421 |
399 |
1.89 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
421 |
399 |
1.89 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2606.8 cm
-1
Scaled (by 0.9479) Zero Point Vibrational Energy (zpe) 2471.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/TZVP
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
0.996 |
| Si2 |
0.000 |
0.000 |
-0.996 |
| H3 |
0.000 |
0.000 |
2.452 |
| H4 |
0.000 |
0.000 |
-2.452 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 1.9921 | 1.4558 | 3.4479 |
Si2 | 1.9921 | | 3.4479 | 1.4558 | H3 | 1.4558 | 3.4479 | | 4.9037 | H4 | 3.4479 | 1.4558 | 4.9037 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
S1C4
Energy calculated at MP2/TZVP
| | hartrees |
|---|
| Energy at 0K | -579.090501 |
| Energy at 298.15K | |
| HF Energy | -578.889167 |
| Nuclear repulsion energy | 63.886958 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2261 |
2143 |
0.00 |
512.08 |
0.26 |
0.41 |
| 2 |
Ag |
633 |
600 |
0.00 |
714.04 |
0.32 |
0.48 |
| 3 |
Ag |
592 |
562 |
0.00 |
3192.89 |
0.40 |
0.57 |
| 4 |
Au |
964 |
913 |
677.66 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2269 |
2151 |
122.60 |
0.00 |
0.30 |
0.47 |
| 6 |
Bu |
481 |
456 |
36.18 |
0.00 |
0.34 |
0.51 |
Unscaled Zero Point Vibrational Energy (zpe) 3599.9 cm
-1
Scaled (by 0.9479) Zero Point Vibrational Energy (zpe) 3412.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/TZVP
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.055 |
0.000 |
| Si2 |
0.000 |
-1.055 |
0.000 |
| H3 |
1.214 |
1.910 |
0.000 |
| H4 |
-1.214 |
-1.910 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1096 | 1.4846 | 3.2034 |
Si2 | 2.1096 | | 3.2034 | 1.4846 | H3 | 1.4846 | 3.2034 | | 4.5257 | H4 | 3.2034 | 1.4846 | 4.5257 | |
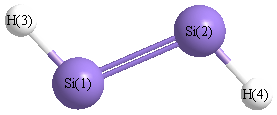 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
125.164 |
|
Si2 |
Si1 |
H3 |
125.164 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S1C4
Energy calculated at MP2/TZVP
| | hartrees |
|---|
| Energy at 0K | -579.118545 |
| Energy at 298.15K | |
| HF Energy | -578.929979 |
| Nuclear repulsion energy | 64.636804 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1684 |
1596 |
3.37 |
98.21 |
0.07 |
0.12 |
| 2 |
A1 |
946 |
896 |
47.68 |
20.80 |
0.38 |
0.55 |
| 3 |
A1 |
527 |
500 |
1.24 |
55.10 |
0.38 |
0.55 |
| 4 |
A2 |
1227 |
1163 |
0.00 |
4.88 |
0.75 |
0.86 |
| 5 |
B1 |
1599 |
1516 |
11.59 |
37.55 |
0.75 |
0.86 |
| 6 |
B2 |
1278 |
1212 |
453.46 |
1.64 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3630.6 cm
-1
Scaled (by 0.9479) Zero Point Vibrational Energy (zpe) 3441.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/TZVP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.112 |
-0.051 |
| Si2 |
0.000 |
-1.112 |
-0.051 |
| H3 |
0.987 |
0.000 |
0.708 |
| H4 |
-0.987 |
0.000 |
0.708 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.2249 | 1.6693 | 1.6693 |
Si2 | 2.2249 | | 1.6693 | 1.6693 | H3 | 1.6693 | 1.6693 | | 1.9733 | H4 | 1.6693 | 1.6693 | 1.9733 | |
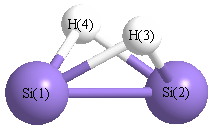 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
48.210 |
|
Si2 |
Si1 |
H3 |
48.210 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S1C3
Energy calculated at MP2/TZVP
| | hartrees |
|---|
| Energy at 0K | -579.102921 |
| Energy at 298.15K | |
| HF Energy | -578.907673 |
| Nuclear repulsion energy | 64.751633 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2260 |
2142 |
99.00 |
347.56 |
0.39 |
0.56 |
| 2 |
A' |
1708 |
1619 |
69.48 |
105.52 |
0.24 |
0.39 |
| 3 |
A' |
1216 |
1153 |
229.05 |
6.37 |
0.65 |
0.79 |
| 4 |
A' |
615 |
583 |
15.89 |
26.61 |
0.39 |
0.57 |
| 5 |
A' |
482 |
457 |
5.76 |
4.56 |
0.30 |
0.47 |
| 6 |
A" |
254 |
240 |
23.73 |
0.12 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3267.4 cm
-1
Scaled (by 0.9479) Zero Point Vibrational Energy (zpe) 3097.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/TZVP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.061 |
-1.151 |
0.000 |
| Si2 |
0.061 |
0.983 |
0.000 |
| H3 |
-1.229 |
-0.022 |
0.000 |
| H4 |
-0.480 |
2.365 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1336 | 1.7141 | 3.5567 |
Si2 | 2.1336 | | 1.6350 | 1.4838 | H3 | 1.7141 | 1.6350 | | 2.5013 | H4 | 3.5567 | 1.4838 | 2.5013 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
158.624 |
|
Si2 |
Si1 |
H3 |
48.808 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability