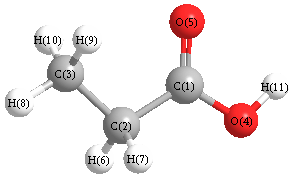
Vibrational Frequencies calculated at MP2/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3793 |
3567 |
71.15 |
|
|
|
| 2 |
A' |
3230 |
3038 |
17.51 |
|
|
|
| 3 |
A' |
3135 |
2949 |
3.44 |
|
|
|
| 4 |
A' |
3135 |
2949 |
24.79 |
|
|
|
| 5 |
A' |
1812 |
1705 |
254.00 |
|
|
|
| 6 |
A' |
1548 |
1456 |
9.10 |
|
|
|
| 7 |
A' |
1510 |
1420 |
12.41 |
|
|
|
| 8 |
A' |
1467 |
1380 |
2.28 |
|
|
|
| 9 |
A' |
1439 |
1353 |
63.04 |
|
|
|
| 10 |
A' |
1312 |
1234 |
16.49 |
|
|
|
| 11 |
A' |
1181 |
1111 |
197.12 |
|
|
|
| 12 |
A' |
1112 |
1046 |
121.22 |
|
|
|
| 13 |
A' |
1041 |
979 |
1.37 |
|
|
|
| 14 |
A' |
835 |
786 |
5.67 |
|
|
|
| 15 |
A' |
612 |
576 |
17.90 |
|
|
|
| 16 |
A' |
471 |
443 |
23.03 |
|
|
|
| 17 |
A' |
253 |
238 |
2.28 |
|
|
|
| 18 |
A" |
3238 |
3046 |
17.32 |
|
|
|
| 19 |
A" |
3185 |
2996 |
2.39 |
|
|
|
| 20 |
A" |
1539 |
1448 |
7.19 |
|
|
|
| 21 |
A" |
1308 |
1230 |
0.02 |
|
|
|
| 22 |
A" |
1134 |
1067 |
0.16 |
|
|
|
| 23 |
A" |
826 |
777 |
9.63 |
|
|
|
| 24 |
A" |
645 |
606 |
102.89 |
|
|
|
| 25 |
A" |
520 |
489 |
29.06 |
|
|
|
| 26 |
A" |
221 |
208 |
0.00 |
|
|
|
| 27 |
A" |
39 |
37 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20269.6 cm
-1
Scaled (by 0.9406) Zero Point Vibrational Energy (zpe) 19065.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.