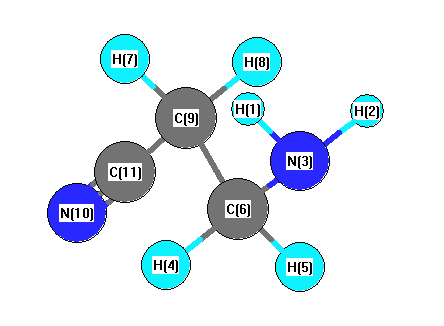
Vibrational Frequencies calculated at MP2/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3555 |
3379 |
0.30 |
|
|
|
| 2 |
A' |
3109 |
2955 |
24.49 |
|
|
|
| 3 |
A' |
3091 |
2937 |
3.61 |
|
|
|
| 4 |
A' |
2198 |
2088 |
0.05 |
|
|
|
| 5 |
A' |
1742 |
1656 |
37.14 |
|
|
|
| 6 |
A' |
1531 |
1455 |
6.74 |
|
|
|
| 7 |
A' |
1511 |
1436 |
3.35 |
|
|
|
| 8 |
A' |
1416 |
1346 |
13.16 |
|
|
|
| 9 |
A' |
1319 |
1253 |
0.78 |
|
|
|
| 10 |
A' |
1146 |
1089 |
7.48 |
|
|
|
| 11 |
A' |
1052 |
1000 |
55.70 |
|
|
|
| 12 |
A' |
963 |
915 |
12.35 |
|
|
|
| 13 |
A' |
879 |
835 |
218.16 |
|
|
|
| 14 |
A' |
520 |
494 |
1.66 |
|
|
|
| 15 |
A' |
381 |
362 |
7.62 |
|
|
|
| 16 |
A' |
162 |
154 |
6.45 |
|
|
|
| 17 |
A" |
3655 |
3473 |
0.81 |
|
|
|
| 18 |
A" |
3171 |
3014 |
19.97 |
|
|
|
| 19 |
A" |
3142 |
2986 |
0.21 |
|
|
|
| 20 |
A" |
1427 |
1356 |
0.32 |
|
|
|
| 21 |
A" |
1342 |
1275 |
0.55 |
|
|
|
| 22 |
A" |
1188 |
1129 |
0.04 |
|
|
|
| 23 |
A" |
994 |
944 |
0.59 |
|
|
|
| 24 |
A" |
764 |
726 |
1.43 |
|
|
|
| 25 |
A" |
376 |
357 |
0.26 |
|
|
|
| 26 |
A" |
313 |
297 |
53.13 |
|
|
|
| 27 |
A" |
110 |
105 |
1.24 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20527.6 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 19507.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.