Jump to
S1C2
Energy calculated at MP2/6-311G*
| | hartrees |
|---|
| Energy at 0K | -749.924889 |
| Energy at 298.15K | |
| HF Energy | -748.185819 |
| Nuclear repulsion energy | 561.734943 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1853 |
1761 |
0.00 |
|
|
|
| 2 |
Ag |
1456 |
1383 |
0.00 |
|
|
|
| 3 |
Ag |
1355 |
1287 |
0.00 |
|
|
|
| 4 |
Ag |
1202 |
1143 |
0.00 |
|
|
|
| 5 |
Ag |
721 |
685 |
0.00 |
|
|
|
| 6 |
Ag |
593 |
563 |
0.00 |
|
|
|
| 7 |
Ag |
382 |
363 |
0.00 |
|
|
|
| 8 |
Ag |
356 |
338 |
0.00 |
|
|
|
| 9 |
Ag |
226 |
214 |
0.00 |
|
|
|
| 10 |
Au |
565 |
537 |
3.45 |
|
|
|
| 11 |
Au |
349 |
331 |
4.10 |
|
|
|
| 12 |
Au |
111 |
105 |
0.04 |
|
|
|
| 13 |
Au |
31i |
30i |
0.12 |
|
|
|
| 14 |
Bg |
621 |
590 |
0.00 |
|
|
|
| 15 |
Bg |
480 |
456 |
0.00 |
|
|
|
| 16 |
Bg |
181 |
172 |
0.00 |
|
|
|
| 17 |
Bu |
1788 |
1699 |
358.69 |
|
|
|
| 18 |
Bu |
1373 |
1304 |
445.24 |
|
|
|
| 19 |
Bu |
1233 |
1172 |
274.59 |
|
|
|
| 20 |
Bu |
967 |
919 |
263.87 |
|
|
|
| 21 |
Bu |
628 |
597 |
5.35 |
|
|
|
| 22 |
Bu |
501 |
477 |
4.11 |
|
|
|
| 23 |
Bu |
292 |
278 |
5.96 |
|
|
|
| 24 |
Bu |
140 |
133 |
0.79 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8669.7 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 8238.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/6-311G*
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.154 |
1.862 |
0.000 |
| C2 |
0.471 |
0.558 |
0.000 |
| C3 |
-0.471 |
-0.558 |
0.000 |
| C4 |
-0.154 |
-1.862 |
0.000 |
| F5 |
1.065 |
2.806 |
0.000 |
| F6 |
-1.065 |
2.342 |
0.000 |
| F7 |
1.773 |
0.226 |
0.000 |
| F8 |
-1.773 |
-0.226 |
0.000 |
| F9 |
1.065 |
-2.342 |
0.000 |
| F10 |
-1.065 |
-2.806 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3420 | 2.4987 | 3.7362 | 1.3122 | 1.3105 | 2.3011 | 2.8410 | 4.3009 | 4.8245 |
C2 | 1.3420 | | 1.4596 | 2.4987 | 2.3258 | 2.3542 | 1.3434 | 2.3764 | 2.9595 | 3.6979 | C3 | 2.4987 | 1.4596 | | 1.3420 | 3.6979 | 2.9595 | 2.3764 | 1.3434 | 2.3542 | 2.3258 | C4 | 3.7362 | 2.4987 | 1.3420 | | 4.8245 | 4.3009 | 2.8410 | 2.3011 | 1.3105 | 1.3122 | F5 | 1.3122 | 2.3258 | 3.6979 | 4.8245 | | 2.1805 | 2.6754 | 4.1530 | 5.1478 | 6.0031 | F6 | 1.3105 | 2.3542 | 2.9595 | 4.3009 | 2.1805 | | 3.5397 | 2.6633 | 5.1450 | 5.1478 | F7 | 2.3011 | 1.3434 | 2.3764 | 2.8410 | 2.6754 | 3.5397 | | 3.5741 | 2.6633 | 4.1530 | F8 | 2.8410 | 2.3764 | 1.3434 | 2.3011 | 4.1530 | 2.6633 | 3.5741 | | 3.5397 | 2.6754 | F9 | 4.3009 | 2.9595 | 2.3542 | 1.3105 | 5.1478 | 5.1450 | 2.6633 | 3.5397 | | 2.1805 | F10 | 4.8245 | 3.6979 | 2.3258 | 1.3122 | 6.0031 | 5.1478 | 4.1530 | 2.6754 | 2.1805 | |
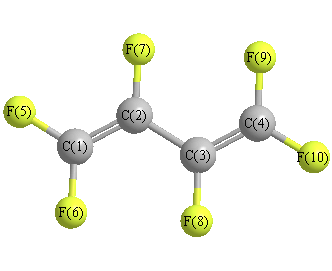 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
126.176 |
|
C1 |
C2 |
F7 |
117.936 |
| C2 |
C1 |
F5 |
122.388 |
|
C2 |
C1 |
F6 |
125.126 |
| C2 |
C3 |
C4 |
126.176 |
|
C2 |
C3 |
F8 |
115.888 |
| C3 |
C2 |
F7 |
115.888 |
|
C3 |
C4 |
F9 |
125.126 |
| C3 |
C4 |
F10 |
122.388 |
|
C4 |
C3 |
F8 |
117.936 |
| F5 |
C1 |
F6 |
112.485 |
|
F9 |
C4 |
F10 |
112.485 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/6-311G*
| | hartrees |
|---|
| Energy at 0K | -749.927758 |
| Energy at 298.15K | -749.928944 |
| HF Energy | -748.187676 |
| Nuclear repulsion energy | 565.935359 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1847 |
1755 |
68.34 |
|
|
|
| 2 |
A |
1433 |
1362 |
22.16 |
|
|
|
| 3 |
A |
1370 |
1302 |
163.26 |
|
|
|
| 4 |
A |
1162 |
1104 |
280.17 |
|
|
|
| 5 |
A |
721 |
685 |
0.15 |
|
|
|
| 6 |
A |
660 |
627 |
1.60 |
|
|
|
| 7 |
A |
545 |
517 |
0.01 |
|
|
|
| 8 |
A |
479 |
455 |
0.32 |
|
|
|
| 9 |
A |
385 |
366 |
0.91 |
|
|
|
| 10 |
A |
261 |
248 |
0.29 |
|
|
|
| 11 |
A |
187 |
178 |
0.39 |
|
|
|
| 12 |
A |
98 |
93 |
0.24 |
|
|
|
| 13 |
A |
47 |
45 |
0.00 |
|
|
|
| 14 |
B |
1817 |
1727 |
238.26 |
|
|
|
| 15 |
B |
1366 |
1298 |
301.98 |
|
|
|
| 16 |
B |
1224 |
1163 |
116.38 |
|
|
|
| 17 |
B |
988 |
939 |
180.01 |
|
|
|
| 18 |
B |
634 |
602 |
5.99 |
|
|
|
| 19 |
B |
622 |
591 |
3.27 |
|
|
|
| 20 |
B |
567 |
539 |
3.84 |
|
|
|
| 21 |
B |
426 |
405 |
3.71 |
|
|
|
| 22 |
B |
296 |
282 |
4.40 |
|
|
|
| 23 |
B |
211 |
200 |
3.48 |
|
|
|
| 24 |
B |
108 |
102 |
0.28 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8725.1 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 8291.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/6-311G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.172 |
1.575 |
-0.372 |
| C2 |
0.169 |
0.707 |
0.587 |
| C3 |
-0.169 |
-0.707 |
0.587 |
| C4 |
0.172 |
-1.575 |
-0.372 |
| F5 |
0.160 |
2.842 |
-0.359 |
| F6 |
-0.846 |
1.242 |
-1.447 |
| F7 |
0.846 |
1.148 |
1.663 |
| F8 |
-0.846 |
-1.148 |
1.663 |
| F9 |
0.846 |
-1.242 |
-1.447 |
| F10 |
-0.160 |
-2.842 |
-0.359 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3383 | 2.4758 | 3.1690 | 1.3097 | 1.3113 | 2.3153 | 3.4652 | 3.1827 | 4.4169 |
C2 | 1.3383 | | 1.4539 | 2.4758 | 2.3349 | 2.3357 | 1.3448 | 2.3718 | 2.8980 | 3.6875 | C3 | 2.4758 | 1.4539 | | 1.3383 | 3.6875 | 2.8980 | 2.3718 | 1.3448 | 2.3357 | 2.3349 | C4 | 3.1690 | 2.4758 | 1.3383 | | 4.4169 | 3.1827 | 3.4652 | 2.3153 | 1.3113 | 1.3097 | F5 | 1.3097 | 2.3349 | 3.6875 | 4.4169 | | 2.1807 | 2.7249 | 4.5839 | 4.2819 | 5.6925 | F6 | 1.3113 | 2.3357 | 2.8980 | 3.1827 | 2.1807 | | 3.5413 | 3.9220 | 3.0057 | 4.2819 | F7 | 2.3153 | 1.3448 | 2.3718 | 3.4652 | 2.7249 | 3.5413 | | 2.8513 | 3.9220 | 4.5839 | F8 | 3.4652 | 2.3718 | 1.3448 | 2.3153 | 4.5839 | 3.9220 | 2.8513 | | 3.5413 | 2.7249 | F9 | 3.1827 | 2.8980 | 2.3357 | 1.3113 | 4.2819 | 3.0057 | 3.9220 | 3.5413 | | 2.1807 | F10 | 4.4169 | 3.6875 | 2.3349 | 1.3097 | 5.6925 | 4.2819 | 4.5839 | 2.7249 | 2.1807 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
124.871 |
|
C1 |
C2 |
F7 |
119.292 |
| C2 |
C1 |
F5 |
123.709 |
|
C2 |
C1 |
F6 |
123.651 |
| C2 |
C3 |
C4 |
124.871 |
|
C2 |
C3 |
F8 |
115.817 |
| C3 |
C2 |
F7 |
115.817 |
|
C3 |
C4 |
F9 |
123.651 |
| C3 |
C4 |
F10 |
123.709 |
|
C4 |
C3 |
F8 |
119.292 |
| F5 |
C1 |
F6 |
112.617 |
|
F9 |
C4 |
F10 |
112.617 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability