Jump to
S1C2
Energy calculated at MP2/6-311G*
| | hartrees |
|---|
| Energy at 0K | -134.729430 |
| Energy at 298.15K | -134.737671 |
| HF Energy | -134.275405 |
| Nuclear repulsion energy | 82.801117 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3536 |
3360 |
1.46 |
|
|
|
| 2 |
A' |
3155 |
2998 |
40.09 |
|
|
|
| 3 |
A' |
3095 |
2941 |
34.77 |
|
|
|
| 4 |
A' |
3065 |
2912 |
21.16 |
|
|
|
| 5 |
A' |
1744 |
1657 |
33.68 |
|
|
|
| 6 |
A' |
1544 |
1467 |
5.36 |
|
|
|
| 7 |
A' |
1525 |
1449 |
0.14 |
|
|
|
| 8 |
A' |
1442 |
1370 |
13.40 |
|
|
|
| 9 |
A' |
1407 |
1337 |
11.74 |
|
|
|
| 10 |
A' |
1188 |
1129 |
12.52 |
|
|
|
| 11 |
A' |
1106 |
1051 |
33.17 |
|
|
|
| 12 |
A' |
936 |
889 |
160.47 |
|
|
|
| 13 |
A' |
900 |
856 |
39.41 |
|
|
|
| 14 |
A' |
408 |
388 |
7.46 |
|
|
|
| 15 |
A" |
3632 |
3452 |
0.00 |
|
|
|
| 16 |
A" |
3164 |
3006 |
61.23 |
|
|
|
| 17 |
A" |
3137 |
2981 |
1.21 |
|
|
|
| 18 |
A" |
1535 |
1459 |
11.10 |
|
|
|
| 19 |
A" |
1427 |
1356 |
0.00 |
|
|
|
| 20 |
A" |
1304 |
1239 |
0.07 |
|
|
|
| 21 |
A" |
1031 |
980 |
1.09 |
|
|
|
| 22 |
A" |
787 |
748 |
0.81 |
|
|
|
| 23 |
A" |
319 |
303 |
42.60 |
|
|
|
| 24 |
A" |
263 |
250 |
14.40 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20825.3 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 19790.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/6-311G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.308 |
-0.089 |
0.000 |
| C2 |
0.000 |
0.572 |
0.000 |
| C3 |
1.214 |
-0.353 |
0.000 |
| H4 |
2.154 |
0.209 |
0.000 |
| H5 |
1.211 |
-0.998 |
0.883 |
| H6 |
1.211 |
-0.998 |
-0.883 |
| H7 |
0.032 |
1.228 |
-0.875 |
| H8 |
0.032 |
1.228 |
0.875 |
| H9 |
-1.385 |
-0.682 |
0.816 |
| H10 |
-1.385 |
-0.682 |
-0.816 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4653 | 2.5351 | 3.4750 | 2.8201 | 2.8201 | 2.0723 | 2.0723 | 1.0122 | 1.0122 |
C2 | 1.4653 | | 1.5258 | 2.1848 | 2.1711 | 2.1711 | 1.0938 | 1.0938 | 2.0391 | 2.0391 | C3 | 2.5351 | 1.5258 | | 1.0958 | 1.0941 | 1.0941 | 2.1586 | 2.1586 | 2.7436 | 2.7436 | H4 | 3.4750 | 2.1848 | 1.0958 | | 1.7685 | 1.7685 | 2.5115 | 2.5115 | 3.7401 | 3.7401 | H5 | 2.8201 | 2.1711 | 1.0941 | 1.7685 | | 1.7668 | 3.0720 | 2.5193 | 2.6162 | 3.1192 | H6 | 2.8201 | 2.1711 | 1.0941 | 1.7685 | 1.7668 | | 2.5193 | 3.0720 | 3.1192 | 2.6162 | H7 | 2.0723 | 1.0938 | 2.1586 | 2.5115 | 3.0720 | 2.5193 | | 1.7493 | 2.9184 | 2.3793 | H8 | 2.0723 | 1.0938 | 2.1586 | 2.5115 | 2.5193 | 3.0720 | 1.7493 | | 2.3793 | 2.9184 | H9 | 1.0122 | 2.0391 | 2.7436 | 3.7401 | 2.6162 | 3.1192 | 2.9184 | 2.3793 | | 1.6327 | H10 | 1.0122 | 2.0391 | 2.7436 | 3.7401 | 3.1192 | 2.6162 | 2.3793 | 2.9184 | 1.6327 | |
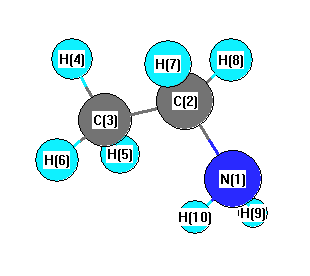 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
115.880 |
|
N1 |
C2 |
H7 |
107.261 |
| N1 |
C2 |
H8 |
107.261 |
|
C2 |
N1 |
H9 |
109.422 |
| C2 |
N1 |
H10 |
109.422 |
|
C2 |
C3 |
H4 |
111.853 |
| C2 |
C3 |
H5 |
110.852 |
|
C2 |
C3 |
H6 |
110.852 |
| C3 |
C2 |
H7 |
109.886 |
|
C3 |
C2 |
H8 |
109.886 |
| H4 |
C3 |
H5 |
107.712 |
|
H4 |
C3 |
H6 |
107.712 |
| H5 |
C3 |
H6 |
107.684 |
|
H7 |
C2 |
H8 |
106.194 |
| H9 |
N1 |
H10 |
107.506 |
|
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/6-311G*
| | hartrees |
|---|
| Energy at 0K | -134.729141 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/6-311G*
Geometric Data calculated at MP2/6-311G*
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability