Jump to
S1C2
S1C3
Energy calculated at MP2/3-21G*
| | hartrees |
|---|
| Energy at 0K | -1583.087161 |
| Energy at 298.15K | |
| HF Energy | -1582.600162 |
| Nuclear repulsion energy | 349.153881 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
490 |
466 |
0.00 |
40.50 |
0.06 |
0.12 |
| 2 |
A1 |
206 |
196 |
0.00 |
3.41 |
0.75 |
0.86 |
| 3 |
B1 |
433 |
412 |
0.00 |
25.43 |
0.75 |
0.86 |
| 4 |
B2 |
330 |
314 |
0.46 |
10.70 |
0.75 |
0.86 |
| 5 |
E |
401 |
381 |
0.00 |
6.93 |
0.75 |
0.86 |
| 5 |
E |
401 |
381 |
0.00 |
6.93 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1130.3 cm
-1
Scaled (by 0.9513) Zero Point Vibrational Energy (zpe) 1075.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/3-21G*
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.436 |
0.315 |
| S2 |
0.000 |
-1.436 |
0.315 |
| S3 |
-1.436 |
0.000 |
-0.315 |
| S4 |
1.436 |
0.000 |
-0.315 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.8723 | 2.1265 | 2.1265 |
S2 | 2.8723 | | 2.1265 | 2.1265 | S3 | 2.1265 | 2.1265 | | 2.8723 | S4 | 2.1265 | 2.1265 | 2.8723 | |
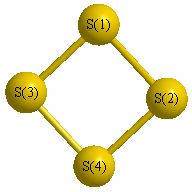 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
47.517 |
|
S1 |
S3 |
S4 |
47.517 |
| S2 |
S1 |
S3 |
47.517 |
|
S2 |
S4 |
S3 |
47.517 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP2/3-21G*
| | hartrees |
|---|
| Energy at 0K | -1583.171168 |
| Energy at 298.15K | |
| HF Energy | -1582.540921 |
| Nuclear repulsion energy | 316.479615 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
657 |
625 |
0.00 |
25.37 |
0.41 |
0.58 |
| 2 |
A1 |
294 |
280 |
0.05 |
0.01 |
0.33 |
0.49 |
| 3 |
A1 |
154 |
146 |
0.00 |
4819.84 |
0.32 |
0.49 |
| 4 |
A2 |
195 |
185 |
0.00 |
0.00 |
0.75 |
0.86 |
| 5 |
B2 |
721 |
686 |
81.51 |
0.00 |
0.75 |
0.86 |
| 6 |
B2 |
270 |
257 |
0.00 |
24.49 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1145.1 cm
-1
Scaled (by 0.9513) Zero Point Vibrational Energy (zpe) 1089.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/3-21G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
1.406 |
0.963 |
| S2 |
0.000 |
-1.406 |
0.963 |
| S3 |
0.000 |
1.406 |
-0.963 |
| S4 |
0.000 |
-1.406 |
-0.963 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.8115 | 1.9266 | 3.4085 |
S2 | 2.8115 | | 3.4085 | 1.9266 | S3 | 1.9266 | 3.4085 | | 2.8119 | S4 | 3.4085 | 1.9266 | 2.8119 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
90.006 |
|
S1 |
S3 |
S4 |
89.994 |
| S2 |
S1 |
S3 |
90.006 |
|
S2 |
S4 |
S3 |
89.994 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP2/3-21G*
| | hartrees |
|---|
| Energy at 0K | -1583.171168 |
| Energy at 298.15K | |
| HF Energy | -1582.540920 |
| Nuclear repulsion energy | 316.474737 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
657 |
625 |
0.00 |
25.40 |
0.41 |
0.58 |
| 2 |
Ag |
153 |
146 |
0.00 |
4749.40 |
0.32 |
0.49 |
| 3 |
Au |
195 |
185 |
0.00 |
0.00 |
0.00 |
0.00 |
| 4 |
B1u |
721 |
686 |
80.38 |
0.00 |
0.75 |
0.86 |
| 5 |
B2u |
294 |
279 |
0.05 |
0.00 |
0.30 |
0.46 |
| 6 |
B3g |
270 |
257 |
0.00 |
24.40 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 1144.9 cm
-1
Scaled (by 0.9513) Zero Point Vibrational Energy (zpe) 1089.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/3-21G*
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.963 |
1.406 |
| S2 |
0.000 |
0.963 |
-1.406 |
| S3 |
0.000 |
-0.963 |
1.406 |
| S4 |
0.000 |
-0.963 |
-1.406 |
Atom - Atom Distances (Å)
| |
S1 |
S2 |
S3 |
S4 |
| S1 | | 2.8122 | 1.9264 | 3.4087 |
S2 | 2.8122 | | 3.4087 | 1.9264 | S3 | 1.9264 | 3.4087 | | 2.8122 | S4 | 3.4087 | 1.9264 | 2.8122 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
S2 |
S4 |
90.000 |
|
S1 |
S3 |
S4 |
90.000 |
| S2 |
S1 |
S3 |
90.000 |
|
S2 |
S4 |
S3 |
90.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability