Jump to
S1C2
S1C3
Energy calculated at MP4=FULL/cc-pCVTZ
| | hartrees |
|---|
| Energy at 0K | -100.327661 |
| Energy at 298.15K | -100.326715 |
| HF Energy | -99.807908 |
| Nuclear repulsion energy | 27.406044 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP4=FULL/cc-pCVTZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.073 |
| C2 |
0.000 |
0.000 |
-0.156 |
| N3 |
0.000 |
0.000 |
1.023 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9168 | 3.0957 |
C2 | 1.9168 | | 1.1789 | N3 | 3.0957 | 1.1789 | |
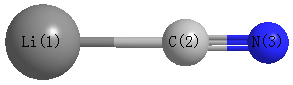 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP4=FULL/cc-pCVTZ
| | hartrees |
|---|
| Energy at 0K | -100.331233 |
| Energy at 298.15K | -100.330506 |
| HF Energy | -99.814496 |
| Nuclear repulsion energy | 29.141707 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP4=FULL/cc-pCVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.397 |
-0.640 |
0.000 |
| C2 |
-0.698 |
-0.372 |
0.000 |
| N3 |
0.000 |
0.593 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.1121 | 1.8632 |
C2 | 2.1121 | | 1.1907 | N3 | 1.8632 | 1.1907 | |
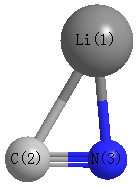 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
61.408 |
|
Li1 |
N3 |
C2 |
84.457 |
| C2 |
Li1 |
N3 |
34.135 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP4=FULL/cc-pCVTZ
| | hartrees |
|---|
| Energy at 0K | -100.330725 |
| Energy at 298.15K | -100.329576 |
| HF Energy | -99.819356 |
| Nuclear repulsion energy | 28.205364 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP4=FULL/cc-pCVTZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.884 |
| C2 |
0.000 |
0.000 |
-1.075 |
| N3 |
0.000 |
0.000 |
0.113 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9590 | 1.7710 |
C2 | 2.9590 | | 1.1881 | N3 | 1.7710 | 1.1881 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability