Jump to
S1C2
Energy calculated at QCISD/6-31G*
| | hartrees |
|---|
| Energy at 0K | -311.259202 |
| Energy at 298.15K | -311.274823 |
| HF Energy | -310.211721 |
| Nuclear repulsion energy | 314.243324 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3142 |
2992 |
46.02 |
|
|
|
| 2 |
A1 |
3076 |
2929 |
41.30 |
|
|
|
| 3 |
A1 |
3061 |
2915 |
18.31 |
|
|
|
| 4 |
A1 |
2998 |
2855 |
76.85 |
|
|
|
| 5 |
A1 |
1590 |
1514 |
0.19 |
|
|
|
| 6 |
A1 |
1560 |
1486 |
6.78 |
|
|
|
| 7 |
A1 |
1546 |
1473 |
0.02 |
|
|
|
| 8 |
A1 |
1501 |
1429 |
0.52 |
|
|
|
| 9 |
A1 |
1468 |
1398 |
1.85 |
|
|
|
| 10 |
A1 |
1376 |
1311 |
2.03 |
|
|
|
| 11 |
A1 |
1209 |
1151 |
2.33 |
|
|
|
| 12 |
A1 |
1102 |
1050 |
5.86 |
|
|
|
| 13 |
A1 |
1036 |
987 |
17.45 |
|
|
|
| 14 |
A1 |
950 |
904 |
7.07 |
|
|
|
| 15 |
A1 |
448 |
427 |
0.81 |
|
|
|
| 16 |
A1 |
325 |
310 |
0.24 |
|
|
|
| 17 |
A1 |
107 |
102 |
0.18 |
|
|
|
| 18 |
A2 |
3135 |
2986 |
0.00 |
|
|
|
| 19 |
A2 |
3113 |
2964 |
0.00 |
|
|
|
| 20 |
A2 |
3023 |
2879 |
0.00 |
|
|
|
| 21 |
A2 |
1551 |
1477 |
0.00 |
|
|
|
| 22 |
A2 |
1346 |
1281 |
0.00 |
|
|
|
| 23 |
A2 |
1299 |
1237 |
0.00 |
|
|
|
| 24 |
A2 |
1213 |
1155 |
0.00 |
|
|
|
| 25 |
A2 |
921 |
877 |
0.00 |
|
|
|
| 26 |
A2 |
782 |
744 |
0.00 |
|
|
|
| 27 |
A2 |
242 |
230 |
0.00 |
|
|
|
| 28 |
A2 |
138 |
131 |
0.00 |
|
|
|
| 29 |
A2 |
72 |
68 |
0.00 |
|
|
|
| 30 |
B1 |
3136 |
2986 |
125.52 |
|
|
|
| 31 |
B1 |
3113 |
2964 |
4.70 |
|
|
|
| 32 |
B1 |
3023 |
2879 |
102.91 |
|
|
|
| 33 |
B1 |
1551 |
1477 |
13.27 |
|
|
|
| 34 |
B1 |
1349 |
1285 |
0.20 |
|
|
|
| 35 |
B1 |
1307 |
1244 |
2.32 |
|
|
|
| 36 |
B1 |
1240 |
1181 |
5.48 |
|
|
|
| 37 |
B1 |
935 |
890 |
3.23 |
|
|
|
| 38 |
B1 |
785 |
747 |
2.26 |
|
|
|
| 39 |
B1 |
245 |
233 |
0.40 |
|
|
|
| 40 |
B1 |
165 |
157 |
6.07 |
|
|
|
| 41 |
B1 |
56 |
54 |
0.26 |
|
|
|
| 42 |
B2 |
3142 |
2992 |
21.46 |
|
|
|
| 43 |
B2 |
3075 |
2929 |
1.78 |
|
|
|
| 44 |
B2 |
3061 |
2915 |
36.65 |
|
|
|
| 45 |
B2 |
2987 |
2844 |
9.50 |
|
|
|
| 46 |
B2 |
1573 |
1498 |
9.15 |
|
|
|
| 47 |
B2 |
1558 |
1484 |
0.67 |
|
|
|
| 48 |
B2 |
1546 |
1472 |
2.48 |
|
|
|
| 49 |
B2 |
1468 |
1398 |
0.01 |
|
|
|
| 50 |
B2 |
1455 |
1385 |
49.46 |
|
|
|
| 51 |
B2 |
1358 |
1293 |
21.54 |
|
|
|
| 52 |
B2 |
1199 |
1142 |
201.16 |
|
|
|
| 53 |
B2 |
1159 |
1104 |
3.72 |
|
|
|
| 54 |
B2 |
1087 |
1035 |
0.50 |
|
|
|
| 55 |
B2 |
925 |
880 |
1.18 |
|
|
|
| 56 |
B2 |
524 |
499 |
5.45 |
|
|
|
| 57 |
B2 |
260 |
247 |
0.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 43302.4 cm
-1
Scaled (by 0.9523) Zero Point Vibrational Energy (zpe) 41236.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.390 |
| C2 |
0.000 |
1.179 |
-0.399 |
| C3 |
0.000 |
-1.179 |
-0.399 |
| C4 |
0.000 |
2.379 |
0.534 |
| C5 |
0.000 |
-2.379 |
0.534 |
| C6 |
0.000 |
3.706 |
-0.227 |
| C7 |
0.000 |
-3.706 |
-0.227 |
| H8 |
0.890 |
1.201 |
-1.055 |
| H9 |
-0.890 |
1.201 |
-1.055 |
| H10 |
-0.890 |
-1.201 |
-1.055 |
| H11 |
0.890 |
-1.201 |
-1.055 |
| H12 |
0.881 |
2.310 |
1.186 |
| H13 |
-0.881 |
2.310 |
1.186 |
| H14 |
-0.881 |
-2.310 |
1.186 |
| H15 |
0.881 |
-2.310 |
1.186 |
| H16 |
0.000 |
4.557 |
0.465 |
| H17 |
-0.887 |
3.796 |
-0.868 |
| H18 |
0.887 |
3.796 |
-0.868 |
| H19 |
0.000 |
-4.557 |
0.465 |
| H20 |
0.887 |
-3.796 |
-0.868 |
| H21 |
-0.887 |
-3.796 |
-0.868 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4195 | 1.4195 | 2.3830 | 2.3830 | 3.7566 | 3.7566 | 2.0794 | 2.0794 | 2.0794 | 2.0794 | 2.5967 | 2.5967 | 2.5967 | 2.5967 | 4.5572 | 4.0958 | 4.0958 | 4.5572 | 4.0958 | 4.0958 |
C2 | 1.4195 | | 2.3588 | 1.5198 | 3.6785 | 2.5320 | 4.8880 | 1.1055 | 1.1055 | 2.6245 | 2.6245 | 2.1369 | 2.1369 | 3.9321 | 3.9321 | 3.4860 | 2.8018 | 2.8018 | 5.8007 | 5.0750 | 5.0750 | C3 | 1.4195 | 2.3588 | | 3.6785 | 1.5198 | 4.8880 | 2.5320 | 2.6245 | 2.6245 | 1.1055 | 1.1055 | 3.9321 | 3.9321 | 2.1369 | 2.1369 | 5.8007 | 5.0750 | 5.0750 | 3.4860 | 2.8018 | 2.8018 | C4 | 2.3830 | 1.5198 | 3.6785 | | 4.7573 | 1.5296 | 6.1316 | 2.1691 | 2.1691 | 4.0164 | 4.0164 | 1.0982 | 1.0982 | 4.8146 | 4.8146 | 2.1790 | 2.1816 | 2.1816 | 6.9355 | 6.3931 | 6.3931 | C5 | 2.3830 | 3.6785 | 1.5198 | 4.7573 | | 6.1316 | 1.5296 | 4.0164 | 4.0164 | 2.1691 | 2.1691 | 4.8146 | 4.8146 | 1.0982 | 1.0982 | 6.9355 | 6.3931 | 6.3931 | 2.1790 | 2.1816 | 2.1816 | C6 | 3.7566 | 2.5320 | 4.8880 | 1.5296 | 6.1316 | | 7.4111 | 2.7840 | 2.7840 | 5.0549 | 5.0549 | 2.1726 | 2.1726 | 6.2412 | 6.2412 | 1.0965 | 1.0981 | 1.0981 | 8.2910 | 7.5805 | 7.5805 | C7 | 3.7566 | 4.8880 | 2.5320 | 6.1316 | 1.5296 | 7.4111 | | 5.0549 | 5.0549 | 2.7840 | 2.7840 | 6.2412 | 6.2412 | 2.1726 | 2.1726 | 8.2910 | 7.5805 | 7.5805 | 1.0965 | 1.0981 | 1.0981 | H8 | 2.0794 | 1.1055 | 2.6245 | 2.1691 | 4.0164 | 2.7840 | 5.0549 | | 1.7791 | 2.9892 | 2.4020 | 2.5001 | 3.0638 | 4.5257 | 4.1648 | 3.7897 | 3.1501 | 2.6013 | 6.0210 | 5.0001 | 5.3063 | H9 | 2.0794 | 1.1055 | 2.6245 | 2.1691 | 4.0164 | 2.7840 | 5.0549 | 1.7791 | | 2.4020 | 2.9892 | 3.0638 | 2.5001 | 4.1648 | 4.5257 | 3.7897 | 2.6013 | 3.1501 | 6.0210 | 5.3063 | 5.0001 | H10 | 2.0794 | 2.6245 | 1.1055 | 4.0164 | 2.1691 | 5.0549 | 2.7840 | 2.9892 | 2.4020 | | 1.7791 | 4.5257 | 4.1648 | 2.5001 | 3.0638 | 6.0210 | 5.0001 | 5.3063 | 3.7897 | 3.1501 | 2.6013 | H11 | 2.0794 | 2.6245 | 1.1055 | 4.0164 | 2.1691 | 5.0549 | 2.7840 | 2.4020 | 2.9892 | 1.7791 | | 4.1648 | 4.5257 | 3.0638 | 2.5001 | 6.0210 | 5.3063 | 5.0001 | 3.7897 | 2.6013 | 3.1501 | H12 | 2.5967 | 2.1369 | 3.9321 | 1.0982 | 4.8146 | 2.1726 | 6.2412 | 2.5001 | 3.0638 | 4.5257 | 4.1648 | | 1.7628 | 4.9440 | 4.6191 | 2.5190 | 3.0905 | 2.5345 | 6.9599 | 6.4411 | 6.6794 | H13 | 2.5967 | 2.1369 | 3.9321 | 1.0982 | 4.8146 | 2.1726 | 6.2412 | 3.0638 | 2.5001 | 4.1648 | 4.5257 | 1.7628 | | 4.6191 | 4.9440 | 2.5190 | 2.5345 | 3.0905 | 6.9599 | 6.6794 | 6.4411 | H14 | 2.5967 | 3.9321 | 2.1369 | 4.8146 | 1.0982 | 6.2412 | 2.1726 | 4.5257 | 4.1648 | 2.5001 | 3.0638 | 4.9440 | 4.6191 | | 1.7628 | 6.9599 | 6.4411 | 6.6794 | 2.5190 | 3.0905 | 2.5345 | H15 | 2.5967 | 3.9321 | 2.1369 | 4.8146 | 1.0982 | 6.2412 | 2.1726 | 4.1648 | 4.5257 | 3.0638 | 2.5001 | 4.6191 | 4.9440 | 1.7628 | | 6.9599 | 6.6794 | 6.4411 | 2.5190 | 2.5345 | 3.0905 | H16 | 4.5572 | 3.4860 | 5.8007 | 2.1790 | 6.9355 | 1.0965 | 8.2910 | 3.7897 | 3.7897 | 6.0210 | 6.0210 | 2.5190 | 2.5190 | 6.9599 | 6.9599 | | 1.7724 | 1.7724 | 9.1131 | 8.5041 | 8.5041 | H17 | 4.0958 | 2.8018 | 5.0750 | 2.1816 | 6.3931 | 1.0981 | 7.5805 | 3.1501 | 2.6013 | 5.0001 | 5.3063 | 3.0905 | 2.5345 | 6.4411 | 6.6794 | 1.7724 | | 1.7741 | 8.5041 | 7.7956 | 7.5911 | H18 | 4.0958 | 2.8018 | 5.0750 | 2.1816 | 6.3931 | 1.0981 | 7.5805 | 2.6013 | 3.1501 | 5.3063 | 5.0001 | 2.5345 | 3.0905 | 6.6794 | 6.4411 | 1.7724 | 1.7741 | | 8.5041 | 7.5911 | 7.7956 | H19 | 4.5572 | 5.8007 | 3.4860 | 6.9355 | 2.1790 | 8.2910 | 1.0965 | 6.0210 | 6.0210 | 3.7897 | 3.7897 | 6.9599 | 6.9599 | 2.5190 | 2.5190 | 9.1131 | 8.5041 | 8.5041 | | 1.7724 | 1.7724 | H20 | 4.0958 | 5.0750 | 2.8018 | 6.3931 | 2.1816 | 7.5805 | 1.0981 | 5.0001 | 5.3063 | 3.1501 | 2.6013 | 6.4411 | 6.6794 | 3.0905 | 2.5345 | 8.5041 | 7.7956 | 7.5911 | 1.7724 | | 1.7741 | H21 | 4.0958 | 5.0750 | 2.8018 | 6.3931 | 2.1816 | 7.5805 | 1.0981 | 5.3063 | 5.0001 | 2.6013 | 3.1501 | 6.6794 | 6.4411 | 2.5345 | 3.0905 | 8.5041 | 7.5911 | 7.7956 | 1.7724 | 1.7741 | |
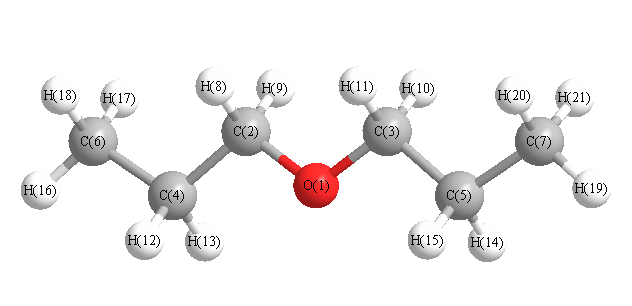 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.289 |
|
O1 |
C2 |
H8 |
110.268 |
| O1 |
C2 |
H9 |
110.268 |
|
O1 |
C3 |
C5 |
108.289 |
| O1 |
C3 |
H10 |
110.268 |
|
O1 |
C3 |
H11 |
110.268 |
| C2 |
O1 |
C3 |
112.380 |
|
C2 |
C4 |
C6 |
112.271 |
| C2 |
C4 |
H12 |
108.344 |
|
C2 |
C4 |
H13 |
108.344 |
| C3 |
C5 |
C7 |
112.271 |
|
C3 |
C5 |
H14 |
108.344 |
| C3 |
C5 |
H15 |
108.344 |
|
C4 |
C2 |
H8 |
110.431 |
| C4 |
C2 |
H9 |
110.431 |
|
C4 |
C6 |
H16 |
111.078 |
| C4 |
C6 |
H17 |
111.188 |
|
C4 |
C6 |
H18 |
111.188 |
| C5 |
C3 |
H10 |
110.431 |
|
C5 |
C3 |
H11 |
110.431 |
| C5 |
C7 |
H19 |
111.078 |
|
C5 |
C7 |
H20 |
111.188 |
| C5 |
C7 |
H21 |
111.188 |
|
C6 |
C4 |
H12 |
110.465 |
| C6 |
C4 |
H13 |
110.465 |
|
C7 |
C5 |
H14 |
110.465 |
| C7 |
C5 |
H15 |
110.465 |
|
H8 |
C2 |
H9 |
107.161 |
| H10 |
C3 |
H11 |
107.161 |
|
H12 |
C4 |
H13 |
106.762 |
| H14 |
C5 |
H15 |
106.762 |
|
H16 |
C6 |
H17 |
107.728 |
| H16 |
C6 |
H18 |
107.728 |
|
H17 |
C6 |
H18 |
107.764 |
| H19 |
C7 |
H20 |
107.728 |
|
H19 |
C7 |
H21 |
107.728 |
| H20 |
C7 |
H21 |
107.764 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/6-31G*
| | hartrees |
|---|
| Energy at 0K | -311.260756 |
| Energy at 298.15K | -311.276605 |
| HF Energy | -310.212115 |
| Nuclear repulsion energy | 325.182813 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3164 |
3013 |
7.55 |
|
|
|
| 2 |
A |
3134 |
2984 |
55.87 |
|
|
|
| 3 |
A |
3105 |
2957 |
11.46 |
|
|
|
| 4 |
A |
3066 |
2920 |
0.39 |
|
|
|
| 5 |
A |
3062 |
2916 |
12.59 |
|
|
|
| 6 |
A |
3029 |
2885 |
3.37 |
|
|
|
| 7 |
A |
3000 |
2857 |
83.06 |
|
|
|
| 8 |
A |
1586 |
1510 |
0.70 |
|
|
|
| 9 |
A |
1560 |
1485 |
4.81 |
|
|
|
| 10 |
A |
1546 |
1472 |
5.99 |
|
|
|
| 11 |
A |
1536 |
1462 |
0.57 |
|
|
|
| 12 |
A |
1491 |
1420 |
2.53 |
|
|
|
| 13 |
A |
1465 |
1395 |
1.48 |
|
|
|
| 14 |
A |
1421 |
1353 |
0.24 |
|
|
|
| 15 |
A |
1345 |
1281 |
2.18 |
|
|
|
| 16 |
A |
1295 |
1233 |
0.00 |
|
|
|
| 17 |
A |
1218 |
1160 |
6.39 |
|
|
|
| 18 |
A |
1178 |
1121 |
9.50 |
|
|
|
| 19 |
A |
1121 |
1067 |
5.87 |
|
|
|
| 20 |
A |
981 |
934 |
7.29 |
|
|
|
| 21 |
A |
946 |
901 |
7.06 |
|
|
|
| 22 |
A |
930 |
886 |
0.55 |
|
|
|
| 23 |
A |
774 |
737 |
0.42 |
|
|
|
| 24 |
A |
493 |
470 |
0.04 |
|
|
|
| 25 |
A |
345 |
328 |
0.43 |
|
|
|
| 26 |
A |
279 |
266 |
0.52 |
|
|
|
| 27 |
A |
166 |
158 |
0.07 |
|
|
|
| 28 |
A |
117 |
112 |
0.02 |
|
|
|
| 29 |
A |
46 |
44 |
0.00 |
|
|
|
| 30 |
B |
3164 |
3013 |
46.67 |
|
|
|
| 31 |
B |
3134 |
2984 |
34.16 |
|
|
|
| 32 |
B |
3105 |
2957 |
33.47 |
|
|
|
| 33 |
B |
3066 |
2919 |
30.31 |
|
|
|
| 34 |
B |
3062 |
2916 |
89.37 |
|
|
|
| 35 |
B |
3028 |
2884 |
92.75 |
|
|
|
| 36 |
B |
2990 |
2847 |
8.20 |
|
|
|
| 37 |
B |
1568 |
1493 |
4.55 |
|
|
|
| 38 |
B |
1559 |
1484 |
10.62 |
|
|
|
| 39 |
B |
1545 |
1472 |
6.37 |
|
|
|
| 40 |
B |
1536 |
1462 |
2.76 |
|
|
|
| 41 |
B |
1466 |
1396 |
10.99 |
|
|
|
| 42 |
B |
1439 |
1371 |
35.97 |
|
|
|
| 43 |
B |
1412 |
1344 |
34.14 |
|
|
|
| 44 |
B |
1337 |
1273 |
1.20 |
|
|
|
| 45 |
B |
1307 |
1244 |
5.67 |
|
|
|
| 46 |
B |
1222 |
1164 |
11.78 |
|
|
|
| 47 |
B |
1202 |
1145 |
139.40 |
|
|
|
| 48 |
B |
1144 |
1090 |
17.54 |
|
|
|
| 49 |
B |
1056 |
1006 |
32.80 |
|
|
|
| 50 |
B |
961 |
915 |
1.15 |
|
|
|
| 51 |
B |
908 |
864 |
0.14 |
|
|
|
| 52 |
B |
807 |
769 |
1.82 |
|
|
|
| 53 |
B |
512 |
488 |
1.41 |
|
|
|
| 54 |
B |
330 |
314 |
0.64 |
|
|
|
| 55 |
B |
236 |
224 |
2.86 |
|
|
|
| 56 |
B |
170 |
162 |
4.91 |
|
|
|
| 57 |
B |
78 |
74 |
1.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 43369.4 cm
-1
Scaled (by 0.9523) Zero Point Vibrational Energy (zpe) 41300.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.114 |
| C2 |
0.022 |
1.182 |
0.902 |
| C3 |
-0.022 |
-1.182 |
0.902 |
| C4 |
0.000 |
2.380 |
-0.034 |
| C5 |
0.000 |
-2.380 |
-0.034 |
| C6 |
-1.265 |
2.435 |
-0.892 |
| C7 |
1.265 |
-2.435 |
-0.892 |
| H8 |
-0.854 |
1.204 |
1.576 |
| H9 |
0.927 |
1.197 |
1.536 |
| H10 |
0.854 |
-1.204 |
1.576 |
| H11 |
-0.927 |
-1.197 |
1.536 |
| H12 |
0.890 |
2.333 |
-0.676 |
| H13 |
0.090 |
3.293 |
0.572 |
| H14 |
-0.890 |
-2.333 |
-0.676 |
| H15 |
-0.090 |
-3.293 |
0.572 |
| H16 |
-1.255 |
3.309 |
-1.555 |
| H17 |
-1.350 |
1.533 |
-1.507 |
| H18 |
-2.161 |
2.501 |
-0.260 |
| H19 |
1.255 |
-3.309 |
-1.555 |
| H20 |
1.350 |
-1.533 |
-1.507 |
| H21 |
2.161 |
-2.501 |
-0.260 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4203 | 1.4203 | 2.3842 | 2.3842 | 2.9222 | 2.9222 | 2.0773 | 2.0766 | 2.0773 | 2.0766 | 2.6191 | 3.3262 | 2.6191 | 3.3262 | 3.9125 | 2.6079 | 3.3264 | 3.9125 | 2.6079 | 3.3264 |
C2 | 1.4203 | | 2.3635 | 1.5205 | 3.6822 | 2.5385 | 4.2236 | 1.1056 | 1.1046 | 2.6144 | 2.6381 | 2.1379 | 2.1385 | 3.9592 | 4.4884 | 3.4916 | 2.7946 | 2.8033 | 5.2648 | 3.8648 | 4.4144 | C3 | 1.4203 | 2.3635 | | 3.6822 | 1.5205 | 4.2236 | 2.5385 | 2.6144 | 2.6381 | 1.1056 | 1.1046 | 3.9592 | 4.4884 | 2.1379 | 2.1385 | 5.2648 | 3.8648 | 4.4144 | 3.4916 | 2.7946 | 2.8033 | C4 | 2.3842 | 1.5205 | 3.6822 | | 4.7592 | 1.5291 | 5.0510 | 2.1690 | 2.1727 | 4.0201 | 4.0143 | 1.0987 | 1.1001 | 4.8387 | 5.7059 | 2.1796 | 2.1698 | 2.1763 | 6.0203 | 4.3934 | 5.3423 | C5 | 2.3842 | 3.6822 | 1.5205 | 4.7592 | | 5.0510 | 1.5291 | 4.0201 | 4.0143 | 2.1690 | 2.1727 | 4.8387 | 5.7059 | 1.0987 | 1.1001 | 6.0203 | 4.3934 | 5.3423 | 2.1796 | 2.1698 | 2.1763 | C6 | 2.9222 | 2.5385 | 4.2236 | 1.5291 | 5.0510 | | 5.4871 | 2.7883 | 3.4966 | 4.8802 | 4.3813 | 2.1678 | 2.1712 | 4.7872 | 6.0277 | 1.0971 | 1.0948 | 1.0985 | 6.3067 | 4.7915 | 6.0410 | C7 | 2.9222 | 4.2236 | 2.5385 | 5.0510 | 1.5291 | 5.4871 | | 4.8802 | 4.3813 | 2.7883 | 3.4966 | 4.7872 | 6.0277 | 2.1678 | 2.1712 | 6.3067 | 4.7915 | 6.0410 | 1.0971 | 1.0948 | 1.0985 | H8 | 2.0773 | 1.1056 | 2.6144 | 2.1690 | 4.0201 | 2.7883 | 4.8802 | | 1.7813 | 2.9516 | 2.4021 | 3.0644 | 2.5032 | 4.1931 | 4.6706 | 3.7940 | 3.1401 | 2.6008 | 5.8831 | 4.6747 | 5.1173 | H9 | 2.0766 | 1.1046 | 2.6381 | 2.1727 | 4.0143 | 3.4966 | 4.3813 | 1.7813 | | 2.4021 | 3.0279 | 2.4867 | 2.4543 | 4.5448 | 4.7038 | 4.3322 | 3.8149 | 3.8027 | 5.4736 | 4.1099 | 4.2923 | H10 | 2.0773 | 2.6144 | 1.1056 | 4.0201 | 2.1690 | 4.8802 | 2.7883 | 2.9516 | 2.4021 | | 1.7813 | 4.1931 | 4.6706 | 3.0644 | 2.5032 | 5.8831 | 4.6747 | 5.1173 | 3.7940 | 3.1401 | 2.6008 | H11 | 2.0766 | 2.6381 | 1.1046 | 4.0143 | 2.1727 | 4.3813 | 3.4966 | 2.4021 | 3.0279 | 1.7813 | | 4.5448 | 4.7038 | 2.4867 | 2.4543 | 5.4736 | 4.1099 | 4.2923 | 4.3322 | 3.8149 | 3.8027 | H12 | 2.6191 | 2.1379 | 3.9592 | 1.0987 | 4.8387 | 2.1678 | 4.7872 | 3.0644 | 2.4867 | 4.1931 | 4.5448 | | 1.7667 | 4.9939 | 5.8458 | 2.5148 | 2.5192 | 3.0840 | 5.7213 | 3.9811 | 5.0154 | H13 | 3.3262 | 2.1385 | 4.4884 | 1.1001 | 5.7059 | 2.1712 | 6.0277 | 2.5032 | 2.4543 | 4.6706 | 4.7038 | 1.7667 | | 5.8458 | 6.5892 | 2.5162 | 3.0810 | 2.5273 | 7.0333 | 5.4042 | 6.2093 | H14 | 2.6191 | 3.9592 | 2.1379 | 4.8387 | 1.0987 | 4.7872 | 2.1678 | 4.1931 | 4.5448 | 3.0644 | 2.4867 | 4.9939 | 5.8458 | | 1.7667 | 5.7213 | 3.9811 | 5.0154 | 2.5148 | 2.5192 | 3.0840 | H15 | 3.3262 | 4.4884 | 2.1385 | 5.7059 | 1.1001 | 6.0277 | 2.1712 | 4.6706 | 4.7038 | 2.5032 | 2.4543 | 5.8458 | 6.5892 | 1.7667 | | 7.0333 | 5.4042 | 6.2093 | 2.5162 | 3.0810 | 2.5273 | H16 | 3.9125 | 3.4916 | 5.2648 | 2.1796 | 6.0203 | 1.0971 | 6.3067 | 3.7940 | 4.3322 | 5.8831 | 5.4736 | 2.5148 | 2.5162 | 5.7213 | 7.0333 | | 1.7787 | 1.7747 | 7.0773 | 5.4982 | 6.8625 | H17 | 2.6079 | 2.7946 | 3.8648 | 2.1698 | 4.3934 | 1.0948 | 4.7915 | 3.1401 | 3.8149 | 4.6747 | 4.1099 | 2.5192 | 3.0810 | 3.9811 | 5.4042 | 1.7787 | | 1.7745 | 5.4982 | 4.0854 | 5.4913 | H18 | 3.3264 | 2.8033 | 4.4144 | 2.1763 | 5.3423 | 1.0985 | 6.0410 | 2.6008 | 3.8027 | 5.1173 | 4.2923 | 3.0840 | 2.5273 | 5.0154 | 6.2093 | 1.7747 | 1.7745 | | 6.8625 | 5.4913 | 6.6105 | H19 | 3.9125 | 5.2648 | 3.4916 | 6.0203 | 2.1796 | 6.3067 | 1.0971 | 5.8831 | 5.4736 | 3.7940 | 4.3322 | 5.7213 | 7.0333 | 2.5148 | 2.5162 | 7.0773 | 5.4982 | 6.8625 | | 1.7787 | 1.7747 | H20 | 2.6079 | 3.8648 | 2.7946 | 4.3934 | 2.1698 | 4.7915 | 1.0948 | 4.6747 | 4.1099 | 3.1401 | 3.8149 | 3.9811 | 5.4042 | 2.5192 | 3.0810 | 5.4982 | 4.0854 | 5.4913 | 1.7787 | | 1.7745 | H21 | 3.3264 | 4.4144 | 2.8033 | 5.3423 | 2.1763 | 6.0410 | 1.0985 | 5.1173 | 4.2923 | 2.6008 | 3.8027 | 5.0154 | 6.2093 | 3.0840 | 2.5273 | 6.8625 | 5.4913 | 6.6105 | 1.7747 | 1.7745 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.283 |
|
O1 |
C2 |
H8 |
110.027 |
| O1 |
C2 |
H9 |
110.036 |
|
O1 |
C3 |
C5 |
108.283 |
| O1 |
C3 |
H10 |
110.027 |
|
O1 |
C3 |
H11 |
110.036 |
| C2 |
O1 |
C3 |
112.613 |
|
C2 |
C4 |
C6 |
112.694 |
| C2 |
C4 |
H12 |
108.342 |
|
C2 |
C4 |
H13 |
108.313 |
| C3 |
C5 |
C7 |
112.694 |
|
C3 |
C5 |
H14 |
108.342 |
| C3 |
C5 |
H15 |
108.313 |
|
C4 |
C2 |
H8 |
110.368 |
| C4 |
C2 |
H9 |
110.724 |
|
C4 |
C6 |
H16 |
111.120 |
| C4 |
C6 |
H17 |
110.482 |
|
C4 |
C6 |
H18 |
110.776 |
| C5 |
C3 |
H10 |
110.368 |
|
C5 |
C3 |
H11 |
110.724 |
| C5 |
C7 |
H19 |
111.120 |
|
C5 |
C7 |
H20 |
110.482 |
| C5 |
C7 |
H21 |
110.776 |
|
C6 |
C4 |
H12 |
110.094 |
| C6 |
C4 |
H13 |
110.277 |
|
C7 |
C5 |
H14 |
110.094 |
| C7 |
C5 |
H15 |
110.277 |
|
H8 |
C2 |
H9 |
107.407 |
| H10 |
C3 |
H11 |
107.407 |
|
H12 |
C4 |
H13 |
106.927 |
| H14 |
C5 |
H15 |
106.927 |
|
H16 |
C6 |
H17 |
108.482 |
| H16 |
C6 |
H18 |
107.865 |
|
H17 |
C6 |
H18 |
108.006 |
| H19 |
C7 |
H20 |
108.482 |
|
H19 |
C7 |
H21 |
107.865 |
| H20 |
C7 |
H21 |
108.006 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability