Jump to
S1C2
Energy calculated at QCISD/6-31G**
| | hartrees |
|---|
| Energy at 0K | -157.965728 |
| Energy at 298.15K | -157.976448 |
| HF Energy | -157.313718 |
| Nuclear repulsion energy | 130.655286 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
|
Au |
122 |
115 |
0.01 |
|
|
|
|
Au |
234 |
220 |
0.00 |
|
|
|
|
Bu |
265 |
249 |
0.01 |
|
|
|
|
Bg |
270 |
254 |
0.00 |
|
|
|
|
Ag |
434 |
409 |
0.00 |
|
|
|
|
Au |
749 |
705 |
1.87 |
|
|
|
|
Bg |
832 |
783 |
0.00 |
|
|
|
|
Ag |
870 |
819 |
0.00 |
|
|
|
|
Au |
988 |
930 |
0.57 |
|
|
|
|
Bu |
1006 |
947 |
3.62 |
|
|
|
|
Bu |
1058 |
996 |
0.07 |
|
|
|
|
Ag |
1109 |
1044 |
0.00 |
|
|
|
|
Ag |
1208 |
1137 |
0.00 |
|
|
|
|
Bg |
1257 |
1183 |
0.00 |
|
|
|
|
Au |
1333 |
1255 |
0.34 |
|
|
|
|
Bu |
1365 |
1285 |
4.81 |
|
|
|
|
Bg |
1371 |
1291 |
0.00 |
|
|
|
|
Ag |
1454 |
1369 |
0.00 |
|
|
|
|
Bu |
1471 |
1385 |
2.14 |
|
|
|
|
Ag |
1472 |
1386 |
0.00 |
|
|
|
|
Ag |
1546 |
1456 |
0.00 |
|
|
|
|
Bu |
1550 |
1460 |
1.15 |
|
|
|
|
Bg |
1558 |
1467 |
0.00 |
|
|
|
|
Au |
1559 |
1467 |
9.20 |
|
|
|
|
Ag |
1564 |
1472 |
0.00 |
|
|
|
|
Bu |
1570 |
1478 |
3.15 |
|
|
|
|
Ag |
3085 |
2905 |
0.00 |
|
|
|
|
Bu |
3092 |
2910 |
50.76 |
|
|
|
|
Bu |
3098 |
2916 |
67.31 |
|
|
|
|
Ag |
3099 |
2918 |
0.00 |
|
|
|
|
Bg |
3118 |
2935 |
0.00 |
|
|
|
|
Au |
3138 |
2954 |
28.53 |
|
|
|
|
Bg |
3176 |
2990 |
0.00 |
|
|
|
|
Au |
3180 |
2993 |
112.98 |
|
|
|
|
Ag |
3181 |
2995 |
0.00 |
|
|
|
|
Bu |
3182 |
2995 |
80.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29780.9 cm
-1
Scaled (by 0.9414) Zero Point Vibrational Energy (zpe) 28035.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G**
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.422 |
0.638 |
0.000 |
| C2 |
0.422 |
-0.638 |
0.000 |
| C3 |
0.422 |
1.912 |
0.000 |
| C4 |
-0.422 |
-1.912 |
0.000 |
| H5 |
-1.079 |
0.633 |
0.876 |
| H6 |
-1.079 |
0.633 |
-0.876 |
| H7 |
1.079 |
-0.633 |
0.876 |
| H8 |
1.079 |
-0.633 |
-0.876 |
| H9 |
-0.205 |
2.805 |
0.000 |
| H10 |
1.065 |
1.953 |
0.882 |
| H11 |
1.065 |
1.953 |
-0.882 |
| H12 |
0.205 |
-2.805 |
0.000 |
| H13 |
-1.065 |
-1.953 |
0.882 |
| H14 |
-1.065 |
-1.953 |
-0.882 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
| C1 | | 1.5294 | 1.5282 | 2.5494 | 1.0947 | 1.0947 | 2.1525 | 2.1525 | 2.1780 | 2.1717 | 2.1717 | 3.4992 | 2.8107 | 2.8107 |
C2 | 1.5294 | | 2.5494 | 1.5282 | 2.1525 | 2.1525 | 1.0947 | 1.0947 | 3.4992 | 2.8107 | 2.8107 | 2.1780 | 2.1717 | 2.1717 | C3 | 1.5282 | 2.5494 | | 3.9155 | 2.1575 | 2.1575 | 2.7700 | 2.7700 | 1.0910 | 1.0919 | 1.0919 | 4.7216 | 4.2332 | 4.2332 | C4 | 2.5494 | 1.5282 | 3.9155 | | 2.7700 | 2.7700 | 2.1575 | 2.1575 | 4.7216 | 4.2332 | 4.2332 | 1.0910 | 1.0919 | 1.0919 | H5 | 1.0947 | 2.1525 | 2.1575 | 2.7700 | | 1.7517 | 2.5012 | 3.0536 | 2.4998 | 2.5170 | 3.0700 | 3.7725 | 2.5854 | 3.1263 | H6 | 1.0947 | 2.1525 | 2.1575 | 2.7700 | 1.7517 | | 3.0536 | 2.5012 | 2.4998 | 3.0700 | 2.5170 | 3.7725 | 3.1263 | 2.5854 | H7 | 2.1525 | 1.0947 | 2.7700 | 2.1575 | 2.5012 | 3.0536 | | 1.7517 | 3.7725 | 2.5854 | 3.1263 | 2.4998 | 2.5170 | 3.0700 | H8 | 2.1525 | 1.0947 | 2.7700 | 2.1575 | 3.0536 | 2.5012 | 1.7517 | | 3.7725 | 3.1263 | 2.5854 | 2.4998 | 3.0700 | 2.5170 | H9 | 2.1780 | 3.4992 | 1.0910 | 4.7216 | 2.4998 | 2.4998 | 3.7725 | 3.7725 | | 1.7650 | 1.7650 | 5.6247 | 4.9143 | 4.9143 | H10 | 2.1717 | 2.8107 | 1.0919 | 4.2332 | 2.5170 | 3.0700 | 2.5854 | 3.1263 | 1.7650 | | 1.7638 | 4.9143 | 4.4479 | 4.7848 | H11 | 2.1717 | 2.8107 | 1.0919 | 4.2332 | 3.0700 | 2.5170 | 3.1263 | 2.5854 | 1.7650 | 1.7638 | | 4.9143 | 4.7848 | 4.4479 | H12 | 3.4992 | 2.1780 | 4.7216 | 1.0910 | 3.7725 | 3.7725 | 2.4998 | 2.4998 | 5.6247 | 4.9143 | 4.9143 | | 1.7650 | 1.7650 | H13 | 2.8107 | 2.1717 | 4.2332 | 1.0919 | 2.5854 | 3.1263 | 2.5170 | 3.0700 | 4.9143 | 4.4479 | 4.7848 | 1.7650 | | 1.7638 | H14 | 2.8107 | 2.1717 | 4.2332 | 1.0919 | 3.1263 | 2.5854 | 3.0700 | 2.5170 | 4.9143 | 4.7848 | 4.4479 | 1.7650 | 1.7638 | |
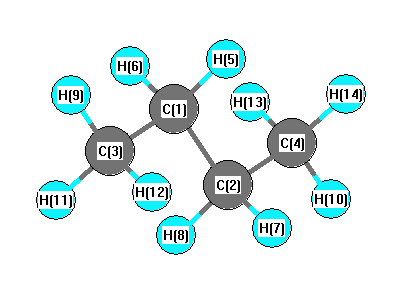 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
112.985 |
|
C1 |
C2 |
H7 |
109.104 |
| C1 |
C2 |
H8 |
109.104 |
|
C1 |
C3 |
H9 |
111.423 |
| C1 |
C3 |
H11 |
110.866 |
|
C1 |
C3 |
H12 |
30.884 |
| C2 |
C1 |
C3 |
112.985 |
|
C2 |
C1 |
H5 |
109.104 |
| C2 |
C1 |
H6 |
109.104 |
|
C2 |
C4 |
H10 |
17.264 |
| C2 |
C4 |
H13 |
110.866 |
|
C2 |
C4 |
H14 |
110.866 |
| C3 |
C1 |
H5 |
109.575 |
|
C3 |
C1 |
H6 |
109.575 |
| C4 |
C2 |
H7 |
109.575 |
|
C4 |
C2 |
H8 |
109.575 |
| H5 |
C1 |
H6 |
106.274 |
|
H7 |
C2 |
H8 |
106.274 |
| H9 |
C3 |
H11 |
107.902 |
|
H9 |
C3 |
H12 |
142.308 |
| H10 |
C4 |
H13 |
94.160 |
|
H10 |
C4 |
H14 |
114.145 |
| H11 |
C3 |
H12 |
93.695 |
|
H13 |
C4 |
H14 |
107.730 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/6-31G**
| | hartrees |
|---|
| Energy at 0K | -157.964633 |
| Energy at 298.15K | |
| Nuclear repulsion energy | 132.514549 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3192 |
3005 |
37.78 |
|
|
|
| 2 |
A |
3178 |
2992 |
0.77 |
|
|
|
| 3 |
A |
3130 |
2947 |
26.75 |
|
|
|
| 4 |
A |
3103 |
2921 |
16.80 |
|
|
|
| 5 |
A |
3091 |
2910 |
30.11 |
|
|
|
| 6 |
A |
1570 |
1478 |
0.78 |
|
|
|
| 7 |
A |
1565 |
1473 |
2.51 |
|
|
|
| 8 |
A |
1547 |
1456 |
0.08 |
|
|
|
| 9 |
A |
1472 |
1386 |
1.62 |
|
|
|
| 10 |
A |
1436 |
1352 |
0.63 |
|
|
|
| 11 |
A |
1354 |
1275 |
0.17 |
|
|
|
| 12 |
A |
1241 |
1168 |
0.07 |
|
|
|
| 13 |
A |
1128 |
1062 |
0.03 |
|
|
|
| 14 |
A |
1025 |
965 |
0.11 |
|
|
|
| 15 |
A |
860 |
810 |
0.00 |
|
|
|
| 16 |
A |
819 |
771 |
0.93 |
|
|
|
| 17 |
A |
328 |
309 |
0.01 |
|
|
|
| 18 |
A |
278 |
262 |
0.01 |
|
|
|
| 19 |
A |
114 |
107 |
0.00 |
|
|
|
| 20 |
B |
3184 |
2997 |
56.98 |
|
|
|
| 21 |
B |
3179 |
2993 |
88.33 |
|
|
|
| 22 |
B |
3136 |
2952 |
32.32 |
|
|
|
| 23 |
B |
3100 |
2918 |
22.83 |
|
|
|
| 24 |
B |
3088 |
2907 |
28.83 |
|
|
|
| 25 |
B |
1565 |
1473 |
6.14 |
|
|
|
| 26 |
B |
1557 |
1466 |
5.35 |
|
|
|
| 27 |
B |
1544 |
1453 |
0.16 |
|
|
|
| 28 |
B |
1475 |
1389 |
4.35 |
|
|
|
| 29 |
B |
1418 |
1335 |
4.26 |
|
|
|
| 30 |
B |
1331 |
1253 |
0.44 |
|
|
|
| 31 |
B |
1194 |
1124 |
1.76 |
|
|
|
| 32 |
B |
1002 |
943 |
0.13 |
|
|
|
| 33 |
B |
994 |
936 |
1.91 |
|
|
|
| 34 |
B |
766 |
721 |
2.23 |
|
|
|
| 35 |
B |
446 |
419 |
0.20 |
|
|
|
| 36 |
B |
223 |
210 |
0.03 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29815.5 cm
-1
Scaled (by 0.9414) Zero Point Vibrational Energy (zpe) 28068.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G**
Point Group is C2h
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability