All results from a given calculation for HCOOC2H5 (Ethyl formate)
using model chemistry: QCISD/Def2TZVPP
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
CS trans |
1A' |
Energy calculated at QCISD/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -267.940327 |
| Energy at 298.15K | |
| HF Energy | -266.940569 |
| Nuclear repulsion energy | 178.187155 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/Def2TZVPP
Geometric Data calculated at QCISD/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.171 |
-0.268 |
0.000 |
| C2 |
-0.689 |
-0.563 |
0.000 |
| O3 |
0.000 |
0.703 |
0.000 |
| C4 |
1.333 |
0.621 |
0.000 |
| O5 |
1.976 |
-0.393 |
0.000 |
| H6 |
-2.731 |
-1.204 |
0.000 |
| H7 |
-2.451 |
0.302 |
0.884 |
| H8 |
-2.451 |
0.302 |
-0.884 |
| H9 |
-0.384 |
-1.126 |
-0.881 |
| H10 |
-0.384 |
-1.126 |
0.881 |
| H11 |
1.760 |
1.628 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5111 | 2.3789 | 3.6159 | 4.1489 | 1.0900 | 1.0892 | 1.0892 | 2.1696 | 2.1696 | 4.3652 |
C2 | 1.5111 | | 1.4418 | 2.3439 | 2.6703 | 2.1399 | 2.1530 | 2.1530 | 1.0890 | 1.0890 | 3.2866 | O3 | 2.3789 | 1.4418 | | 1.3358 | 2.2592 | 3.3310 | 2.6365 | 2.6365 | 2.0663 | 2.0663 | 1.9885 | C4 | 3.6159 | 2.3439 | 1.3358 | | 1.2002 | 4.4554 | 3.8995 | 3.8995 | 2.6034 | 2.6034 | 1.0934 | O5 | 4.1489 | 2.6703 | 2.2592 | 1.2002 | | 4.7760 | 4.5674 | 4.5674 | 2.6230 | 2.6230 | 2.0319 | H6 | 1.0900 | 2.1399 | 3.3310 | 4.4554 | 4.7760 | | 1.7689 | 1.7689 | 2.5082 | 2.5082 | 5.3096 | H7 | 1.0892 | 2.1530 | 2.6365 | 3.8995 | 4.5674 | 1.7689 | | 1.7689 | 3.0709 | 2.5131 | 4.5030 | H8 | 1.0892 | 2.1530 | 2.6365 | 3.8995 | 4.5674 | 1.7689 | 1.7689 | | 2.5131 | 3.0709 | 4.5030 | H9 | 2.1696 | 1.0890 | 2.0663 | 2.6034 | 2.6230 | 2.5082 | 3.0709 | 2.5131 | | 1.7610 | 3.5998 | H10 | 2.1696 | 1.0890 | 2.0663 | 2.6034 | 2.6230 | 2.5082 | 2.5131 | 3.0709 | 1.7610 | | 3.5998 | H11 | 4.3652 | 3.2866 | 1.9885 | 1.0934 | 2.0319 | 5.3096 | 4.5030 | 4.5030 | 3.5998 | 3.5998 | |
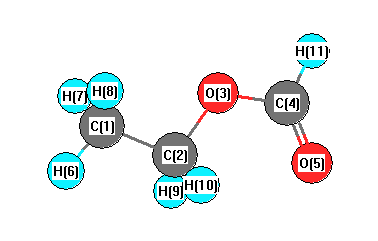 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
107.316 |
|
C1 |
C2 |
H9 |
112.091 |
| C1 |
C2 |
H10 |
112.091 |
|
C2 |
C1 |
H6 |
109.648 |
| C2 |
C1 |
H7 |
110.739 |
|
C2 |
C1 |
H8 |
110.739 |
| C2 |
O3 |
C4 |
115.047 |
|
O3 |
C2 |
H9 |
108.668 |
| O3 |
C2 |
H10 |
108.668 |
|
O3 |
C4 |
O5 |
125.865 |
| O3 |
C4 |
H11 |
109.479 |
|
O5 |
C4 |
H11 |
124.656 |
| H6 |
C1 |
H7 |
108.533 |
|
H6 |
C1 |
H8 |
108.533 |
| H7 |
C1 |
H8 |
108.588 |
|
H9 |
C2 |
H10 |
107.912 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability