Jump to
S1C2
Vibrational Frequencies calculated at QCISD/3-21G*
Geometric Data calculated at QCISD/3-21G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -233.495838 |
| Energy at 298.15K | -233.509327 |
| Nuclear repulsion energy | 251.753764 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3130 |
3008 |
47.85 |
|
|
|
| 2 |
A' |
3103 |
2983 |
14.78 |
|
|
|
| 3 |
A' |
3082 |
2962 |
40.70 |
|
|
|
| 4 |
A' |
3074 |
2954 |
20.06 |
|
|
|
| 5 |
A' |
3069 |
2950 |
13.35 |
|
|
|
| 6 |
A' |
3050 |
2931 |
8.71 |
|
|
|
| 7 |
A' |
3011 |
2894 |
22.25 |
|
|
|
| 8 |
A' |
3006 |
2889 |
21.53 |
|
|
|
| 9 |
A' |
1579 |
1517 |
4.20 |
|
|
|
| 10 |
A' |
1570 |
1509 |
5.17 |
|
|
|
| 11 |
A' |
1566 |
1505 |
1.08 |
|
|
|
| 12 |
A' |
1549 |
1488 |
1.34 |
|
|
|
| 13 |
A' |
1477 |
1419 |
2.11 |
|
|
|
| 14 |
A' |
1460 |
1403 |
9.58 |
|
|
|
| 15 |
A' |
1342 |
1290 |
4.95 |
|
|
|
| 16 |
A' |
1309 |
1258 |
6.86 |
|
|
|
| 17 |
A' |
1262 |
1213 |
0.61 |
|
|
|
| 18 |
A' |
1219 |
1172 |
0.38 |
|
|
|
| 19 |
A' |
1027 |
987 |
0.89 |
|
|
|
| 20 |
A' |
998 |
959 |
0.33 |
|
|
|
| 21 |
A' |
957 |
919 |
0.13 |
|
|
|
| 22 |
A' |
890 |
856 |
0.30 |
|
|
|
| 23 |
A' |
887 |
853 |
1.14 |
|
|
|
| 24 |
A' |
697 |
670 |
1.48 |
|
|
|
| 25 |
A' |
604 |
581 |
0.89 |
|
|
|
| 26 |
A' |
391 |
376 |
0.23 |
|
|
|
| 27 |
A' |
353 |
340 |
0.05 |
|
|
|
| 28 |
A' |
171 |
164 |
0.00 |
|
|
|
| 29 |
A" |
3107 |
2986 |
12.99 |
|
|
|
| 30 |
A" |
3074 |
2954 |
47.45 |
|
|
|
| 31 |
A" |
3068 |
2949 |
1.02 |
|
|
|
| 32 |
A" |
3046 |
2927 |
42.46 |
|
|
|
| 33 |
A" |
1581 |
1520 |
6.40 |
|
|
|
| 34 |
A" |
1564 |
1503 |
0.13 |
|
|
|
| 35 |
A" |
1541 |
1481 |
3.34 |
|
|
|
| 36 |
A" |
1302 |
1251 |
0.63 |
|
|
|
| 37 |
A" |
1287 |
1236 |
0.00 |
|
|
|
| 38 |
A" |
1256 |
1207 |
0.49 |
|
|
|
| 39 |
A" |
1202 |
1155 |
4.17 |
|
|
|
| 40 |
A" |
1122 |
1079 |
0.24 |
|
|
|
| 41 |
A" |
1033 |
993 |
0.02 |
|
|
|
| 42 |
A" |
887 |
852 |
1.42 |
|
|
|
| 43 |
A" |
865 |
831 |
0.00 |
|
|
|
| 44 |
A" |
797 |
766 |
0.14 |
|
|
|
| 45 |
A" |
383 |
368 |
0.25 |
|
|
|
| 46 |
A" |
304 |
292 |
0.01 |
|
|
|
| 47 |
A" |
273 |
262 |
0.01 |
|
|
|
| 48 |
A" |
238 |
229 |
0.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36880.0 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 35445.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.106 |
0.416 |
0.000 |
| C2 |
0.377 |
-1.754 |
0.000 |
| C3 |
-0.106 |
-0.723 |
1.106 |
| C4 |
-0.106 |
-0.723 |
-1.106 |
| C5 |
1.235 |
1.185 |
0.000 |
| C6 |
-1.308 |
1.375 |
0.000 |
| H7 |
1.468 |
-1.863 |
0.000 |
| H8 |
-0.098 |
-2.741 |
0.000 |
| H9 |
0.545 |
-0.567 |
1.975 |
| H10 |
0.545 |
-0.567 |
-1.975 |
| H11 |
-1.130 |
-0.949 |
1.433 |
| H12 |
-1.130 |
-0.949 |
-1.433 |
| H13 |
2.085 |
0.487 |
0.000 |
| H14 |
-2.251 |
0.808 |
0.000 |
| H15 |
1.308 |
1.823 |
-0.894 |
| H16 |
1.308 |
1.823 |
0.894 |
| H17 |
-1.286 |
2.019 |
0.894 |
| H18 |
-1.286 |
2.019 |
-0.894 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.2232 | 1.5880 | 1.5880 | 1.5456 | 1.5383 | 2.7700 | 3.1576 | 2.3005 | 2.3005 | 2.2289 | 2.2289 | 2.1922 | 2.1811 | 2.1858 | 2.1858 | 2.1818 | 2.1818 |
C2 | 2.2232 | | 1.5867 | 1.5867 | 3.0617 | 3.5542 | 1.0968 | 1.0955 | 2.3106 | 2.3106 | 2.2303 | 2.2303 | 2.8176 | 3.6701 | 3.8032 | 3.8032 | 4.2186 | 4.2186 | C3 | 1.5880 | 1.5867 | | 2.2117 | 2.5812 | 2.6597 | 2.2359 | 2.3010 | 1.0969 | 3.1530 | 1.0992 | 2.7475 | 2.7363 | 2.8585 | 3.5335 | 2.9204 | 2.9928 | 3.5933 | C4 | 1.5880 | 1.5867 | 2.2117 | | 2.5812 | 2.6597 | 2.2359 | 2.3010 | 3.1530 | 1.0969 | 2.7475 | 1.0992 | 2.7363 | 2.8585 | 2.9204 | 3.5335 | 3.5933 | 2.9928 | C5 | 1.5456 | 3.0617 | 2.5812 | 2.5812 | | 2.5507 | 3.0570 | 4.1464 | 2.7293 | 2.7293 | 3.4934 | 3.4934 | 1.1003 | 3.5070 | 1.1013 | 1.1013 | 2.8022 | 2.8022 | C6 | 1.5383 | 3.5542 | 2.6597 | 2.6597 | 2.5507 | | 4.2658 | 4.2909 | 3.3330 | 3.3330 | 2.7364 | 2.7364 | 3.5082 | 1.1007 | 2.8009 | 2.8009 | 1.1015 | 1.1015 | H7 | 2.7700 | 1.0968 | 2.2359 | 2.2359 | 3.0570 | 4.2658 | | 1.7955 | 2.5367 | 2.5367 | 3.1054 | 3.1054 | 2.4294 | 4.5792 | 3.7969 | 3.7969 | 4.8429 | 4.8429 | H8 | 3.1576 | 1.0955 | 2.3010 | 2.3010 | 4.1464 | 4.2909 | 1.7955 | | 3.0069 | 3.0069 | 2.5166 | 2.5166 | 3.8968 | 4.1512 | 4.8592 | 4.8592 | 4.9867 | 4.9867 | H9 | 2.3005 | 2.3106 | 1.0969 | 3.1530 | 2.7293 | 3.3330 | 2.5367 | 3.0069 | | 3.9508 | 1.8014 | 3.8171 | 2.7179 | 3.6891 | 3.8123 | 2.7324 | 3.3477 | 4.2745 | H10 | 2.3005 | 2.3106 | 3.1530 | 1.0969 | 2.7293 | 3.3330 | 2.5367 | 3.0069 | 3.9508 | | 3.8171 | 1.8014 | 2.7179 | 3.6891 | 2.7324 | 3.8123 | 4.2745 | 3.3477 | H11 | 2.2289 | 2.2303 | 1.0992 | 2.7475 | 3.4934 | 2.7364 | 3.1054 | 2.5166 | 1.8014 | 3.8171 | | 2.8667 | 3.8021 | 2.5291 | 4.3645 | 3.7310 | 3.0200 | 3.7744 | H12 | 2.2289 | 2.2303 | 2.7475 | 1.0992 | 3.4934 | 2.7364 | 3.1054 | 2.5166 | 3.8171 | 1.8014 | 2.8667 | | 3.8021 | 2.5291 | 3.7310 | 4.3645 | 3.7744 | 3.0200 | H13 | 2.1922 | 2.8176 | 2.7363 | 2.7363 | 1.1003 | 3.5082 | 2.4294 | 3.8968 | 2.7179 | 2.7179 | 3.8021 | 3.8021 | | 4.3486 | 1.7866 | 1.7866 | 3.8098 | 3.8098 | H14 | 2.1811 | 3.6701 | 2.8585 | 2.8585 | 3.5070 | 1.1007 | 4.5792 | 4.1512 | 3.6891 | 3.6891 | 2.5291 | 2.5291 | 4.3486 | | 3.8078 | 3.8078 | 1.7879 | 1.7879 | H15 | 2.1858 | 3.8032 | 3.5335 | 2.9204 | 1.1013 | 2.8009 | 3.7969 | 4.8592 | 3.8123 | 2.7324 | 4.3645 | 3.7310 | 1.7866 | 3.8078 | | 1.7890 | 3.1570 | 2.6015 | H16 | 2.1858 | 3.8032 | 2.9204 | 3.5335 | 1.1013 | 2.8009 | 3.7969 | 4.8592 | 2.7324 | 3.8123 | 3.7310 | 4.3645 | 1.7866 | 3.8078 | 1.7890 | | 2.6015 | 3.1570 | H17 | 2.1818 | 4.2186 | 2.9928 | 3.5933 | 2.8022 | 1.1015 | 4.8429 | 4.9867 | 3.3477 | 4.2745 | 3.0200 | 3.7744 | 3.8098 | 1.7879 | 3.1570 | 2.6015 | | 1.7880 | H18 | 2.1818 | 4.2186 | 3.5933 | 2.9928 | 2.8022 | 1.1015 | 4.8429 | 4.9867 | 4.2745 | 3.3477 | 3.7744 | 3.0200 | 3.8098 | 1.7879 | 2.6015 | 3.1570 | 1.7880 | |
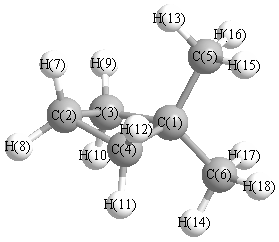 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
88.899 |
|
C1 |
C3 |
H9 |
116.732 |
| C1 |
C3 |
H11 |
110.771 |
|
C1 |
C4 |
C2 |
88.899 |
| C1 |
C4 |
H10 |
116.732 |
|
C1 |
C4 |
H12 |
110.771 |
| C1 |
C5 |
H13 |
110.771 |
|
C1 |
C5 |
H15 |
110.210 |
| C1 |
C5 |
H16 |
110.210 |
|
C1 |
C6 |
H14 |
110.382 |
| C1 |
C6 |
H17 |
110.390 |
|
C1 |
C6 |
H18 |
110.390 |
| C2 |
C3 |
H9 |
117.694 |
|
C2 |
C3 |
H11 |
110.972 |
| C2 |
C4 |
H10 |
117.694 |
|
C2 |
C4 |
H12 |
110.972 |
| C3 |
C1 |
C4 |
88.274 |
|
C3 |
C1 |
C5 |
110.911 |
| C3 |
C1 |
C6 |
116.581 |
|
C3 |
C2 |
C4 |
88.365 |
| C3 |
C2 |
H7 |
111.545 |
|
C3 |
C2 |
H8 |
116.973 |
| C4 |
C1 |
C5 |
110.911 |
|
C4 |
C1 |
C6 |
116.581 |
| C4 |
C2 |
H7 |
111.545 |
|
C4 |
C2 |
H8 |
116.973 |
| C5 |
C1 |
C6 |
111.605 |
|
H7 |
C2 |
H8 |
109.963 |
| H9 |
C3 |
H11 |
110.221 |
|
H10 |
C4 |
H12 |
110.221 |
| H13 |
C5 |
H15 |
108.486 |
|
H13 |
C5 |
H16 |
108.486 |
| H14 |
C6 |
H17 |
108.554 |
|
H14 |
C6 |
H18 |
108.554 |
| H15 |
C5 |
H16 |
108.618 |
|
H17 |
C6 |
H18 |
108.511 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability