Jump to
S1C2
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -355.208679 |
| Energy at 298.15K | -355.213948 |
| HF Energy | -354.538507 |
| Nuclear repulsion energy | 228.878303 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3680 |
3536 |
57.76 |
|
|
|
| 2 |
A' |
3547 |
3409 |
63.87 |
|
|
|
| 3 |
A' |
3500 |
3363 |
25.75 |
|
|
|
| 4 |
A' |
1767 |
1698 |
47.59 |
|
|
|
| 5 |
A' |
1734 |
1667 |
255.92 |
|
|
|
| 6 |
A' |
1678 |
1613 |
160.39 |
|
|
|
| 7 |
A' |
1428 |
1372 |
7.26 |
|
|
|
| 8 |
A' |
1329 |
1278 |
73.76 |
|
|
|
| 9 |
A' |
1135 |
1091 |
185.81 |
|
|
|
| 10 |
A' |
1104 |
1061 |
112.95 |
|
|
|
| 11 |
A' |
738 |
710 |
4.40 |
|
|
|
| 12 |
A' |
607 |
584 |
66.26 |
|
|
|
| 13 |
A' |
534 |
513 |
3.09 |
|
|
|
| 14 |
A' |
423 |
407 |
9.24 |
|
|
|
| 15 |
A' |
268 |
258 |
10.89 |
|
|
|
| 16 |
A" |
827 |
795 |
0.04 |
|
|
|
| 17 |
A" |
653 |
628 |
302.06 |
|
|
|
| 18 |
A" |
626 |
602 |
79.28 |
|
|
|
| 19 |
A" |
575 |
553 |
153.74 |
|
|
|
| 20 |
A" |
427 |
410 |
45.16 |
|
|
|
| 21 |
A" |
111 |
106 |
4.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13345.0 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 12825.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.748 |
0.000 |
| C2 |
-0.048 |
-0.797 |
0.000 |
| O3 |
-1.100 |
-1.455 |
0.000 |
| O4 |
1.045 |
1.407 |
0.000 |
| O5 |
-1.262 |
1.288 |
0.000 |
| N6 |
1.229 |
-1.283 |
0.000 |
| H7 |
1.391 |
-2.283 |
0.000 |
| H8 |
2.007 |
-0.634 |
0.000 |
| H9 |
-1.183 |
2.280 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5455 | 2.4621 | 1.2355 | 1.3726 | 2.3743 | 3.3346 | 2.4371 | 1.9361 |
C2 | 1.5455 | | 1.2404 | 2.4599 | 2.4122 | 1.3671 | 2.0686 | 2.0621 | 3.2798 | O3 | 2.4621 | 1.2404 | | 3.5763 | 2.7476 | 2.3354 | 2.6245 | 3.2138 | 3.7364 | O4 | 1.2355 | 2.4599 | 3.5763 | | 2.3103 | 2.6963 | 3.7056 | 2.2561 | 2.3935 | O5 | 1.3726 | 2.4122 | 2.7476 | 2.3103 | | 3.5801 | 4.4481 | 3.7924 | 0.9958 | N6 | 2.3743 | 1.3671 | 2.3354 | 2.6963 | 3.5801 | | 1.0124 | 1.0134 | 4.3035 | H7 | 3.3346 | 2.0686 | 2.6245 | 3.7056 | 4.4481 | 1.0124 | | 1.7603 | 5.2391 | H8 | 2.4371 | 2.0621 | 3.2138 | 2.2561 | 3.7924 | 1.0134 | 1.7603 | | 4.3213 | H9 | 1.9361 | 3.2798 | 3.7364 | 2.3935 | 0.9958 | 4.3035 | 5.2391 | 4.3213 | |
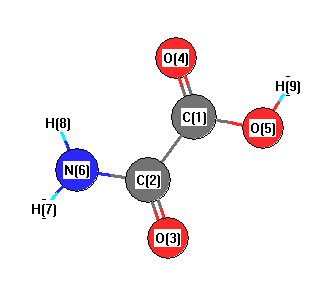 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
123.834 |
|
C1 |
C2 |
N6 |
109.056 |
| C1 |
O5 |
H9 |
108.617 |
|
C2 |
C1 |
O4 |
124.010 |
| C2 |
C1 |
O5 |
111.372 |
|
C2 |
N6 |
H7 |
120.021 |
| C2 |
N6 |
H8 |
119.310 |
|
O3 |
C2 |
N6 |
127.109 |
| O4 |
C1 |
O5 |
124.618 |
|
H7 |
N6 |
H8 |
120.669 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -355.214213 |
| Energy at 298.15K | -355.219702 |
| Nuclear repulsion energy | 230.263519 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3662 |
3519 |
65.73 |
|
|
|
| 2 |
A' |
3532 |
3395 |
77.45 |
|
|
|
| 3 |
A' |
3404 |
3272 |
79.36 |
|
|
|
| 4 |
A' |
1808 |
1737 |
94.01 |
|
|
|
| 5 |
A' |
1737 |
1669 |
243.71 |
|
|
|
| 6 |
A' |
1676 |
1611 |
44.92 |
|
|
|
| 7 |
A' |
1410 |
1355 |
95.76 |
|
|
|
| 8 |
A' |
1341 |
1289 |
451.00 |
|
|
|
| 9 |
A' |
1177 |
1131 |
43.45 |
|
|
|
| 10 |
A' |
1121 |
1077 |
0.18 |
|
|
|
| 11 |
A' |
752 |
723 |
12.80 |
|
|
|
| 12 |
A' |
629 |
604 |
9.78 |
|
|
|
| 13 |
A' |
555 |
533 |
1.48 |
|
|
|
| 14 |
A' |
416 |
400 |
5.30 |
|
|
|
| 15 |
A' |
278 |
267 |
43.18 |
|
|
|
| 16 |
A" |
815 |
784 |
9.15 |
|
|
|
| 17 |
A" |
734 |
706 |
195.71 |
|
|
|
| 18 |
A" |
663 |
637 |
77.78 |
|
|
|
| 19 |
A" |
640 |
615 |
274.38 |
|
|
|
| 20 |
A" |
444 |
427 |
0.90 |
|
|
|
| 21 |
A" |
153 |
147 |
5.96 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13472.3 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 12948.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.024 |
-0.792 |
0.000 |
| C2 |
0.000 |
0.758 |
0.000 |
| O3 |
-1.104 |
1.352 |
0.000 |
| O4 |
1.056 |
-1.462 |
0.000 |
| O5 |
-1.248 |
-1.299 |
0.000 |
| N6 |
1.241 |
1.290 |
0.000 |
| H7 |
1.379 |
2.294 |
0.000 |
| H8 |
2.036 |
0.659 |
0.000 |
| H9 |
-1.869 |
-0.512 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5504 | 2.4231 | 1.2302 | 1.3693 | 2.4119 | 3.3707 | 2.4807 | 1.9134 |
C2 | 1.5504 | | 1.2538 | 2.4580 | 2.4062 | 1.3499 | 2.0642 | 2.0382 | 2.2598 | O3 | 2.4231 | 1.2538 | | 3.5474 | 2.6554 | 2.3453 | 2.6555 | 3.2155 | 2.0156 | O4 | 1.2302 | 2.4580 | 3.5474 | | 2.3099 | 2.7581 | 3.7695 | 2.3359 | 3.0752 | O5 | 1.3693 | 2.4062 | 2.6554 | 2.3099 | | 3.5915 | 4.4512 | 3.8234 | 1.0022 | N6 | 2.4119 | 1.3499 | 2.3453 | 2.7581 | 3.5915 | | 1.0133 | 1.0155 | 3.5942 | H7 | 3.3707 | 2.0642 | 2.6555 | 3.7695 | 4.4512 | 1.0133 | | 1.7623 | 4.2924 | H8 | 2.4807 | 2.0382 | 3.2155 | 2.3359 | 3.8234 | 1.0155 | 1.7623 | | 4.0767 | H9 | 1.9134 | 2.2598 | 2.0156 | 3.0752 | 1.0022 | 3.5942 | 4.2924 | 4.0767 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.181 |
|
C1 |
C2 |
N6 |
112.342 |
| C1 |
O5 |
H9 |
106.549 |
|
C2 |
C1 |
O4 |
123.846 |
| C2 |
C1 |
O5 |
110.843 |
|
C2 |
N6 |
H7 |
121.076 |
| C2 |
N6 |
H8 |
118.328 |
|
O3 |
C2 |
N6 |
128.477 |
| O4 |
C1 |
O5 |
125.311 |
|
H7 |
N6 |
H8 |
120.597 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability