Jump to
S1C2
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -745.028302 |
| Energy at 298.15K | -745.029423 |
| HF Energy | -743.942267 |
| Nuclear repulsion energy | 554.515522 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1859 |
1787 |
0.00 |
|
|
|
| 2 |
Ag |
1445 |
1389 |
0.00 |
|
|
|
| 3 |
Ag |
1350 |
1297 |
0.00 |
|
|
|
| 4 |
Ag |
1204 |
1157 |
0.00 |
|
|
|
| 5 |
Ag |
692 |
665 |
0.00 |
|
|
|
| 6 |
Ag |
584 |
561 |
0.00 |
|
|
|
| 7 |
Ag |
366 |
352 |
0.00 |
|
|
|
| 8 |
Ag |
338 |
325 |
0.00 |
|
|
|
| 9 |
Ag |
223 |
215 |
0.00 |
|
|
|
| 10 |
Au |
583 |
561 |
8.80 |
|
|
|
| 11 |
Au |
348 |
334 |
10.22 |
|
|
|
| 12 |
Au |
119 |
114 |
0.17 |
|
|
|
| 13 |
Au |
14 |
13 |
0.04 |
|
|
|
| 14 |
Bg |
725 |
697 |
0.00 |
|
|
|
| 15 |
Bg |
480 |
461 |
0.00 |
|
|
|
| 16 |
Bg |
181 |
174 |
0.00 |
|
|
|
| 17 |
Bu |
1810 |
1740 |
157.02 |
|
|
|
| 18 |
Bu |
1372 |
1319 |
223.79 |
|
|
|
| 19 |
Bu |
1233 |
1185 |
145.71 |
|
|
|
| 20 |
Bu |
948 |
911 |
157.93 |
|
|
|
| 21 |
Bu |
604 |
580 |
5.94 |
|
|
|
| 22 |
Bu |
476 |
457 |
6.54 |
|
|
|
| 23 |
Bu |
279 |
268 |
8.46 |
|
|
|
| 24 |
Bu |
142 |
137 |
1.91 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8686.2 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 8348.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.165 |
1.843 |
0.000 |
| C2 |
0.480 |
0.547 |
0.000 |
| C3 |
-0.480 |
-0.547 |
0.000 |
| C4 |
-0.165 |
-1.843 |
0.000 |
| F5 |
1.099 |
2.828 |
0.000 |
| F6 |
-1.099 |
2.328 |
0.000 |
| F7 |
1.813 |
0.184 |
0.000 |
| F8 |
-1.813 |
-0.184 |
0.000 |
| F9 |
1.099 |
-2.328 |
0.000 |
| F10 |
-1.099 |
-2.828 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3337 | 2.4745 | 3.6998 | 1.3581 | 1.3538 | 2.3379 | 2.8316 | 4.2737 | 4.8388 |
C2 | 1.3337 | | 1.4542 | 2.4745 | 2.3644 | 2.3801 | 1.3815 | 2.4059 | 2.9403 | 3.7258 | C3 | 2.4745 | 1.4542 | | 1.3337 | 3.7258 | 2.9403 | 2.4059 | 1.3815 | 2.3801 | 2.3644 | C4 | 3.6998 | 2.4745 | 1.3337 | | 4.8388 | 4.2737 | 2.8316 | 2.3379 | 1.3538 | 1.3581 | F5 | 1.3581 | 2.3644 | 3.7258 | 4.8388 | | 2.2543 | 2.7388 | 4.1896 | 5.1561 | 6.0686 | F6 | 1.3538 | 2.3801 | 2.9403 | 4.2737 | 2.2543 | | 3.6157 | 2.6113 | 5.1484 | 5.1561 | F7 | 2.3379 | 1.3815 | 2.4059 | 2.8316 | 2.7388 | 3.6157 | | 3.6440 | 2.6113 | 4.1896 | F8 | 2.8316 | 2.4059 | 1.3815 | 2.3379 | 4.1896 | 2.6113 | 3.6440 | | 3.6157 | 2.7388 | F9 | 4.2737 | 2.9403 | 2.3801 | 1.3538 | 5.1561 | 5.1484 | 2.6113 | 3.6157 | | 2.2543 | F10 | 4.8388 | 3.7258 | 2.3644 | 1.3581 | 6.0686 | 5.1561 | 4.1896 | 2.7388 | 2.2543 | |
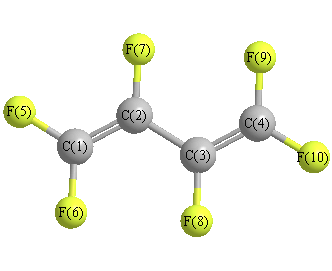 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
125.078 |
|
C1 |
C2 |
F7 |
118.861 |
| C2 |
C1 |
F5 |
122.885 |
|
C2 |
C1 |
F6 |
124.656 |
| C2 |
C3 |
C4 |
125.078 |
|
C2 |
C3 |
F8 |
116.061 |
| C3 |
C2 |
F7 |
116.061 |
|
C3 |
C4 |
F9 |
124.656 |
| C3 |
C4 |
F10 |
122.885 |
|
C4 |
C3 |
F8 |
118.861 |
| F5 |
C1 |
F6 |
112.459 |
|
F9 |
C4 |
F10 |
112.459 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -745.022932 |
| Energy at 298.15K | -745.023943 |
| HF Energy | -743.935995 |
| Nuclear repulsion energy | 559.809056 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1844 |
1772 |
44.00 |
|
|
|
| 2 |
A |
1397 |
1343 |
0.36 |
|
|
|
| 3 |
A |
1373 |
1319 |
105.46 |
|
|
|
| 4 |
A |
1141 |
1096 |
143.77 |
|
|
|
| 5 |
A |
694 |
667 |
0.01 |
|
|
|
| 6 |
A |
671 |
645 |
1.85 |
|
|
|
| 7 |
A |
521 |
501 |
0.06 |
|
|
|
| 8 |
A |
460 |
442 |
1.12 |
|
|
|
| 9 |
A |
369 |
354 |
2.07 |
|
|
|
| 10 |
A |
247 |
238 |
0.14 |
|
|
|
| 11 |
A |
190 |
183 |
0.37 |
|
|
|
| 12 |
A |
104 |
100 |
0.54 |
|
|
|
| 13 |
A |
45 |
43 |
0.01 |
|
|
|
| 14 |
B |
1827 |
1756 |
95.57 |
|
|
|
| 15 |
B |
1366 |
1313 |
164.09 |
|
|
|
| 16 |
B |
1236 |
1188 |
55.32 |
|
|
|
| 17 |
B |
959 |
922 |
97.89 |
|
|
|
| 18 |
B |
602 |
578 |
3.78 |
|
|
|
| 19 |
B |
590 |
567 |
7.24 |
|
|
|
| 20 |
B |
549 |
528 |
5.75 |
|
|
|
| 21 |
B |
390 |
375 |
5.87 |
|
|
|
| 22 |
B |
290 |
278 |
4.51 |
|
|
|
| 23 |
B |
204 |
196 |
6.77 |
|
|
|
| 24 |
B |
112 |
107 |
0.59 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8589.8 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 8255.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.092 |
1.529 |
-0.380 |
| C2 |
0.264 |
0.681 |
0.632 |
| C3 |
-0.264 |
-0.681 |
0.632 |
| C4 |
-0.092 |
-1.529 |
-0.380 |
| F5 |
0.601 |
2.783 |
-0.426 |
| F6 |
-0.601 |
1.205 |
-1.501 |
| F7 |
0.953 |
1.097 |
1.759 |
| F8 |
-0.953 |
-1.097 |
1.759 |
| F9 |
0.601 |
-1.205 |
-1.501 |
| F10 |
-0.601 |
-2.783 |
-0.426 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3314 | 2.4562 | 3.0627 | 1.3539 | 1.3567 | 2.3456 | 3.5440 | 2.9974 | 4.3667 |
C2 | 1.3314 | | 1.4607 | 2.4562 | 2.3768 | 2.3601 | 1.3845 | 2.4314 | 2.8663 | 3.7232 | C3 | 2.4562 | 1.4607 | | 1.3314 | 3.7232 | 2.8663 | 2.4314 | 1.3845 | 2.3601 | 2.3768 | C4 | 3.0627 | 2.4562 | 1.3314 | | 4.3667 | 2.9974 | 3.5440 | 2.3456 | 1.3567 | 1.3539 | F5 | 1.3539 | 2.3768 | 3.7232 | 4.3667 | | 2.2557 | 2.7818 | 4.7154 | 4.1295 | 5.6933 | F6 | 1.3567 | 2.3601 | 2.8663 | 2.9974 | 2.2557 | | 3.6123 | 4.0056 | 2.6920 | 4.1295 | F7 | 2.3456 | 1.3845 | 2.4314 | 3.5440 | 2.7818 | 3.6123 | | 2.9058 | 4.0056 | 4.7154 | F8 | 3.5440 | 2.4314 | 1.3845 | 2.3456 | 4.7154 | 4.0056 | 2.9058 | | 3.6123 | 2.7818 | F9 | 2.9974 | 2.8663 | 2.3601 | 1.3567 | 4.1295 | 2.6920 | 4.0056 | 3.6123 | | 2.2557 | F10 | 4.3667 | 3.7232 | 2.3768 | 1.3539 | 5.6933 | 4.1295 | 4.7154 | 2.7818 | 2.2557 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
123.148 |
|
C1 |
C2 |
F7 |
119.448 |
| C2 |
C1 |
F5 |
124.536 |
|
C2 |
C1 |
F6 |
122.804 |
| C2 |
C3 |
C4 |
123.148 |
|
C2 |
C3 |
F8 |
117.392 |
| C3 |
C2 |
F7 |
117.392 |
|
C3 |
C4 |
F9 |
122.804 |
| C3 |
C4 |
F10 |
124.536 |
|
C4 |
C3 |
F8 |
119.448 |
| F5 |
C1 |
F6 |
112.647 |
|
F9 |
C4 |
F10 |
112.647 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability