Jump to
S1C2
Vibrational Frequencies calculated at QCISD/3-21G*
Geometric Data calculated at QCISD/3-21G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -233.500127 |
| Energy at 298.15K | -233.512338 |
| HF Energy | -232.878700 |
| Nuclear repulsion energy | 228.259631 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3125 |
3003 |
56.39 |
|
|
|
| 2 |
A |
3108 |
2987 |
4.80 |
|
|
|
| 3 |
A |
3094 |
2974 |
38.10 |
|
|
|
| 4 |
A |
3087 |
2967 |
32.38 |
|
|
|
| 5 |
A |
3085 |
2965 |
33.73 |
|
|
|
| 6 |
A |
3071 |
2951 |
1.79 |
|
|
|
| 7 |
A |
3064 |
2945 |
22.15 |
|
|
|
| 8 |
A |
3053 |
2935 |
2.49 |
|
|
|
| 9 |
A |
3025 |
2908 |
26.83 |
|
|
|
| 10 |
A |
3021 |
2903 |
13.34 |
|
|
|
| 11 |
A |
3019 |
2901 |
28.84 |
|
|
|
| 12 |
A |
3014 |
2897 |
10.62 |
|
|
|
| 13 |
A |
1705 |
1639 |
0.14 |
|
|
|
| 14 |
A |
1583 |
1521 |
6.99 |
|
|
|
| 15 |
A |
1578 |
1517 |
6.66 |
|
|
|
| 16 |
A |
1577 |
1515 |
4.39 |
|
|
|
| 17 |
A |
1570 |
1509 |
3.80 |
|
|
|
| 18 |
A |
1568 |
1507 |
1.51 |
|
|
|
| 19 |
A |
1558 |
1497 |
0.55 |
|
|
|
| 20 |
A |
1475 |
1418 |
3.96 |
|
|
|
| 21 |
A |
1473 |
1416 |
3.48 |
|
|
|
| 22 |
A |
1471 |
1414 |
5.87 |
|
|
|
| 23 |
A |
1408 |
1354 |
0.69 |
|
|
|
| 24 |
A |
1371 |
1318 |
0.03 |
|
|
|
| 25 |
A |
1348 |
1296 |
0.42 |
|
|
|
| 26 |
A |
1336 |
1284 |
0.80 |
|
|
|
| 27 |
A |
1306 |
1255 |
0.18 |
|
|
|
| 28 |
A |
1199 |
1152 |
1.35 |
|
|
|
| 29 |
A |
1136 |
1091 |
2.75 |
|
|
|
| 30 |
A |
1113 |
1070 |
0.40 |
|
|
|
| 31 |
A |
1097 |
1055 |
0.82 |
|
|
|
| 32 |
A |
1025 |
985 |
1.91 |
|
|
|
| 33 |
A |
1007 |
968 |
0.37 |
|
|
|
| 34 |
A |
981 |
942 |
3.49 |
|
|
|
| 35 |
A |
922 |
886 |
0.79 |
|
|
|
| 36 |
A |
898 |
863 |
9.63 |
|
|
|
| 37 |
A |
866 |
832 |
3.03 |
|
|
|
| 38 |
A |
761 |
731 |
2.36 |
|
|
|
| 39 |
A |
726 |
698 |
32.97 |
|
|
|
| 40 |
A |
582 |
559 |
6.55 |
|
|
|
| 41 |
A |
475 |
457 |
0.98 |
|
|
|
| 42 |
A |
368 |
354 |
0.02 |
|
|
|
| 43 |
A |
301 |
289 |
0.04 |
|
|
|
| 44 |
A |
243 |
233 |
0.00 |
|
|
|
| 45 |
A |
201 |
193 |
0.01 |
|
|
|
| 46 |
A |
105 |
101 |
0.22 |
|
|
|
| 47 |
A |
93 |
89 |
0.00 |
|
|
|
| 48 |
A |
33 |
31 |
0.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36610.7 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 35186.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
2.423 |
1.006 |
-0.034 |
| C2 |
2.002 |
-0.456 |
-0.176 |
| C3 |
0.798 |
-0.988 |
0.108 |
| C4 |
-0.446 |
-0.253 |
0.604 |
| C5 |
-1.544 |
-0.184 |
-0.503 |
| C6 |
-2.837 |
0.514 |
0.005 |
| H7 |
1.606 |
1.631 |
0.347 |
| H8 |
3.277 |
1.094 |
0.656 |
| H9 |
2.738 |
1.409 |
-1.010 |
| H10 |
2.787 |
-1.120 |
-0.549 |
| H11 |
0.652 |
-2.060 |
-0.059 |
| H12 |
-0.198 |
0.769 |
0.926 |
| H13 |
-0.858 |
-0.782 |
1.479 |
| H14 |
-1.136 |
0.362 |
-1.368 |
| H15 |
-1.779 |
-1.206 |
-0.841 |
| H16 |
-3.599 |
0.557 |
-0.787 |
| H17 |
-3.256 |
-0.036 |
0.861 |
| H18 |
-2.614 |
1.541 |
0.330 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 1.5283 | 2.5765 | 3.1974 | 4.1681 | 5.2835 | 1.0972 | 1.1012 | 1.1013 | 2.2179 | 3.5410 | 2.8015 | 4.0319 | 3.8554 | 4.8169 | 6.0853 | 5.8434 | 5.0787 |
C2 | 1.5283 | | 1.3469 | 2.5771 | 3.5714 | 4.9389 | 2.1875 | 2.1724 | 2.1715 | 1.0938 | 2.0999 | 2.7487 | 3.3208 | 3.4555 | 3.9118 | 5.7245 | 5.3763 | 5.0552 | C3 | 2.5765 | 1.3469 | | 1.5270 | 2.5498 | 3.9340 | 2.7505 | 3.2832 | 3.2802 | 2.0994 | 1.0946 | 2.1785 | 2.1599 | 2.7821 | 2.7545 | 4.7451 | 4.2317 | 4.2525 | C4 | 3.1974 | 2.5771 | 1.5270 | | 1.5610 | 2.5818 | 2.7966 | 3.9588 | 3.9376 | 3.5402 | 2.2151 | 1.0997 | 1.1031 | 2.1781 | 2.1848 | 3.5403 | 2.8307 | 2.8277 | C5 | 4.1681 | 3.5714 | 2.5498 | 1.5610 | | 1.5549 | 3.7328 | 5.1199 | 4.5968 | 4.4313 | 2.9218 | 2.1822 | 2.1820 | 1.1007 | 1.1016 | 2.2029 | 2.1946 | 2.1944 | C6 | 5.2835 | 4.9389 | 3.9340 | 2.5818 | 1.5549 | | 4.5937 | 6.1757 | 5.7375 | 5.8831 | 4.3356 | 2.8070 | 2.7878 | 2.1910 | 2.1888 | 1.0997 | 1.1004 | 1.1004 | H7 | 1.0972 | 2.1875 | 2.7505 | 2.7966 | 3.7328 | 4.5937 | | 1.7820 | 1.7814 | 3.1250 | 3.8331 | 2.0808 | 3.6294 | 3.4741 | 4.5730 | 5.4335 | 5.1653 | 4.2207 | H8 | 1.1012 | 2.1724 | 3.2832 | 3.9588 | 5.1199 | 6.1757 | 1.7820 | | 1.7784 | 2.5678 | 4.1649 | 3.5001 | 4.6144 | 4.9099 | 5.7524 | 7.0456 | 6.6332 | 5.9167 | H9 | 1.1013 | 2.1715 | 3.2802 | 3.9376 | 4.5968 | 5.7375 | 1.7814 | 1.7784 | | 2.5713 | 4.1584 | 3.5747 | 4.8920 | 4.0298 | 5.2225 | 6.3980 | 6.4440 | 5.5193 | H10 | 2.2179 | 1.0938 | 2.0994 | 3.5402 | 4.4313 | 5.8831 | 3.1250 | 2.5678 | 2.5713 | | 2.3842 | 3.8281 | 4.1853 | 4.2735 | 4.5766 | 6.6069 | 6.2999 | 6.0852 | H11 | 3.5410 | 2.0999 | 1.0946 | 2.2151 | 2.9218 | 4.3356 | 3.8331 | 4.1649 | 4.1584 | 2.3842 | | 3.1130 | 2.5051 | 3.2829 | 2.6926 | 5.0444 | 4.4958 | 4.8766 | H12 | 2.8015 | 2.7487 | 2.1785 | 1.0997 | 2.1822 | 2.8070 | 2.0808 | 3.5001 | 3.5747 | 3.8281 | 3.1130 | | 1.7743 | 2.5117 | 3.0856 | 3.8139 | 3.1633 | 2.6057 | H13 | 4.0319 | 3.3208 | 2.1599 | 1.1031 | 2.1820 | 2.7878 | 3.6294 | 4.6144 | 4.8920 | 4.1853 | 2.5051 | 1.7743 | | 3.0816 | 2.5323 | 3.8007 | 2.5869 | 3.1310 | H14 | 3.8554 | 3.4555 | 2.7821 | 2.1781 | 1.1007 | 2.1910 | 3.4741 | 4.9099 | 4.0298 | 4.2735 | 3.2829 | 2.5117 | 3.0816 | | 1.7747 | 2.5374 | 3.1018 | 2.5411 | H15 | 4.8169 | 3.9118 | 2.7545 | 2.1848 | 1.1016 | 2.1888 | 4.5730 | 5.7524 | 5.2225 | 4.5766 | 2.6926 | 3.0856 | 2.5323 | 1.7747 | | 2.5342 | 2.5390 | 3.1004 | H16 | 6.0853 | 5.7245 | 4.7451 | 3.5403 | 2.2029 | 1.0997 | 5.4335 | 7.0456 | 6.3980 | 6.6069 | 5.0444 | 3.8139 | 3.8007 | 2.5374 | 2.5342 | | 1.7850 | 1.7847 | H17 | 5.8434 | 5.3763 | 4.2317 | 2.8307 | 2.1946 | 1.1004 | 5.1653 | 6.6332 | 6.4440 | 6.2999 | 4.4958 | 3.1633 | 2.5869 | 3.1018 | 2.5390 | 1.7850 | | 1.7837 | H18 | 5.0787 | 5.0552 | 4.2525 | 2.8277 | 2.1944 | 1.1004 | 4.2207 | 5.9167 | 5.5193 | 6.0852 | 4.8766 | 2.6057 | 3.1310 | 2.5411 | 3.1004 | 1.7847 | 1.7837 | |
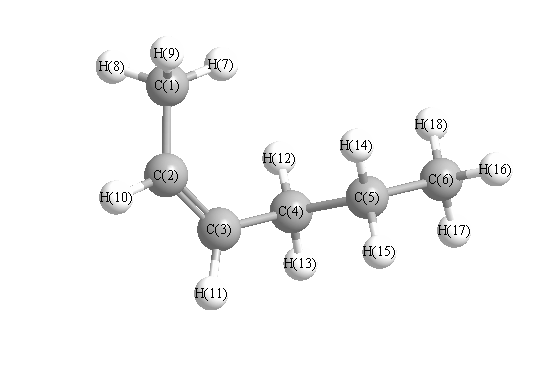 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
127.187 |
|
C1 |
C2 |
H10 |
114.508 |
| C2 |
C1 |
H7 |
111.805 |
|
C2 |
C1 |
H8 |
110.359 |
| C2 |
C1 |
H9 |
110.286 |
|
C2 |
C3 |
C4 |
127.357 |
| C2 |
C3 |
H11 |
118.284 |
|
C3 |
C2 |
H10 |
118.305 |
| C3 |
C4 |
C5 |
111.319 |
|
C3 |
C4 |
H12 |
111.023 |
| C3 |
C4 |
H13 |
109.353 |
|
C4 |
C3 |
H11 |
114.324 |
| C4 |
C5 |
C6 |
111.908 |
|
C4 |
C5 |
H14 |
108.593 |
| C4 |
C5 |
H15 |
109.055 |
|
C5 |
C4 |
H12 |
108.972 |
| C5 |
C4 |
H13 |
108.753 |
|
C5 |
C6 |
H16 |
110.999 |
| C5 |
C6 |
H17 |
110.310 |
|
C5 |
C6 |
H18 |
110.290 |
| C6 |
C5 |
H14 |
110.008 |
|
C6 |
C5 |
H15 |
109.785 |
| H7 |
C1 |
H8 |
108.312 |
|
H7 |
C1 |
H9 |
108.251 |
| H8 |
C1 |
H9 |
107.702 |
|
H12 |
C4 |
H13 |
107.312 |
| H14 |
C5 |
H16 |
94.423 |
|
H16 |
C6 |
H17 |
108.453 |
| H16 |
C6 |
H18 |
108.426 |
|
H17 |
C6 |
H18 |
108.286 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability