Jump to
S1C2
Vibrational Frequencies calculated at QCISD/3-21G*
Geometric Data calculated at QCISD/3-21G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at QCISD/3-21G*
| | hartrees |
|---|
| Energy at 0K | -230.242465 |
| Energy at 298.15K | -230.250814 |
| Nuclear repulsion energy | 173.966412 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3130 |
3008 |
10.67 |
|
|
|
| 2 |
A |
3114 |
2993 |
23.61 |
|
|
|
| 3 |
A |
3107 |
2986 |
20.50 |
|
|
|
| 4 |
A |
3090 |
2969 |
10.82 |
|
|
|
| 5 |
A |
3054 |
2935 |
5.83 |
|
|
|
| 6 |
A |
3043 |
2924 |
15.69 |
|
|
|
| 7 |
A |
3032 |
2914 |
9.64 |
|
|
|
| 8 |
A |
3022 |
2905 |
7.97 |
|
|
|
| 9 |
A |
1701 |
1635 |
40.92 |
|
|
|
| 10 |
A |
1577 |
1516 |
5.82 |
|
|
|
| 11 |
A |
1574 |
1513 |
6.64 |
|
|
|
| 12 |
A |
1559 |
1499 |
11.71 |
|
|
|
| 13 |
A |
1544 |
1484 |
17.39 |
|
|
|
| 14 |
A |
1538 |
1478 |
7.30 |
|
|
|
| 15 |
A |
1483 |
1425 |
7.95 |
|
|
|
| 16 |
A |
1448 |
1391 |
24.19 |
|
|
|
| 17 |
A |
1391 |
1336 |
17.19 |
|
|
|
| 18 |
A |
1335 |
1283 |
0.16 |
|
|
|
| 19 |
A |
1193 |
1147 |
74.14 |
|
|
|
| 20 |
A |
1177 |
1132 |
0.10 |
|
|
|
| 21 |
A |
1125 |
1081 |
6.77 |
|
|
|
| 22 |
A |
1016 |
976 |
3.93 |
|
|
|
| 23 |
A |
1004 |
965 |
1.27 |
|
|
|
| 24 |
A |
942 |
906 |
11.67 |
|
|
|
| 25 |
A |
797 |
766 |
3.48 |
|
|
|
| 26 |
A |
742 |
713 |
1.42 |
|
|
|
| 27 |
A |
581 |
558 |
6.35 |
|
|
|
| 28 |
A |
490 |
471 |
0.01 |
|
|
|
| 29 |
A |
404 |
388 |
5.38 |
|
|
|
| 30 |
A |
257 |
247 |
5.09 |
|
|
|
| 31 |
A |
192 |
185 |
0.08 |
|
|
|
| 32 |
A |
131 |
126 |
0.02 |
|
|
|
| 33 |
A |
69 |
67 |
0.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24930.1 cm
-1
Scaled (by 0.9611) Zero Point Vibrational Energy (zpe) 23960.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/3-21G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.911 |
-0.497 |
0.001 |
| C2 |
0.527 |
0.170 |
-0.000 |
| C3 |
-0.673 |
-0.793 |
-0.001 |
| C4 |
-2.019 |
-0.027 |
0.001 |
| O5 |
0.378 |
1.407 |
-0.000 |
| H6 |
2.683 |
0.281 |
-0.001 |
| H7 |
2.023 |
-1.132 |
0.893 |
| H8 |
2.023 |
-1.135 |
-0.889 |
| H9 |
-2.864 |
-0.729 |
0.000 |
| H10 |
-2.088 |
0.615 |
0.888 |
| H11 |
-2.089 |
0.618 |
-0.885 |
| H12 |
-0.594 |
-1.444 |
-0.887 |
| H13 |
-0.593 |
-1.446 |
0.883 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5365 | 2.6010 | 3.9583 | 2.4450 | 1.0962 | 1.1002 | 1.1002 | 4.7808 | 4.2451 | 4.2462 | 2.8210 | 2.8193 |
C2 | 1.5365 | | 1.5384 | 2.5536 | 1.2462 | 2.1592 | 2.1747 | 2.1749 | 3.5080 | 2.7978 | 2.7977 | 2.1555 | 2.1555 | C3 | 2.6010 | 1.5384 | | 1.5489 | 2.4378 | 3.5239 | 2.8606 | 2.8594 | 2.1920 | 2.1856 | 2.1856 | 1.1025 | 1.1025 | C4 | 3.9583 | 2.5536 | 1.5489 | | 2.7926 | 4.7124 | 4.2844 | 4.2849 | 1.0986 | 1.0976 | 1.0976 | 2.1976 | 2.1977 | O5 | 2.4450 | 1.2462 | 2.4378 | 2.7926 | | 2.5660 | 3.1542 | 3.1556 | 3.8818 | 2.7380 | 2.7368 | 3.1396 | 3.1407 | H6 | 1.0962 | 2.1592 | 3.5239 | 4.7124 | 2.5660 | | 1.7966 | 1.7968 | 5.6385 | 4.8651 | 4.8654 | 3.8078 | 3.8073 | H7 | 1.1002 | 2.1747 | 2.8606 | 4.2844 | 3.1542 | 1.7966 | | 1.7818 | 4.9844 | 4.4670 | 4.8095 | 3.1801 | 2.6346 | H8 | 1.1002 | 2.1749 | 2.8594 | 4.2849 | 3.1556 | 1.7968 | 1.7818 | | 4.9843 | 4.8090 | 4.4702 | 2.6352 | 3.1752 | H9 | 4.7808 | 3.5080 | 2.1920 | 1.0986 | 3.8818 | 5.6385 | 4.9844 | 4.9843 | | 1.7880 | 1.7880 | 2.5406 | 2.5407 | H10 | 4.2451 | 2.7978 | 2.1856 | 1.0976 | 2.7380 | 4.8651 | 4.4670 | 4.8090 | 1.7880 | | 1.7735 | 3.1028 | 2.5471 | H11 | 4.2462 | 2.7977 | 2.1856 | 1.0976 | 2.7368 | 4.8654 | 4.8095 | 4.4702 | 1.7880 | 1.7735 | | 2.5471 | 3.1028 | H12 | 2.8210 | 2.1555 | 1.1025 | 2.1976 | 3.1396 | 3.8078 | 3.1801 | 2.6352 | 2.5406 | 3.1028 | 2.5471 | | 1.7704 | H13 | 2.8193 | 2.1555 | 1.1025 | 2.1977 | 3.1407 | 3.8073 | 2.6346 | 3.1752 | 2.5407 | 2.5471 | 3.1028 | 1.7704 | |
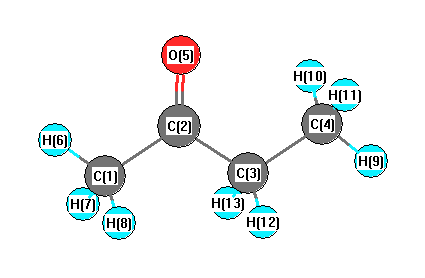 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
115.535 |
|
C1 |
C2 |
O5 |
122.614 |
| C2 |
C1 |
H6 |
109.052 |
|
C2 |
C1 |
H7 |
110.031 |
| C2 |
C1 |
H8 |
110.046 |
|
C2 |
C3 |
C4 |
111.608 |
| C2 |
C3 |
H12 |
108.277 |
|
C2 |
C3 |
H13 |
108.280 |
| C3 |
C2 |
O5 |
121.851 |
|
C3 |
C4 |
H9 |
110.626 |
| C3 |
C4 |
H10 |
110.182 |
|
C3 |
C4 |
H11 |
110.181 |
| C4 |
C3 |
H12 |
110.840 |
|
C4 |
C3 |
H13 |
110.843 |
| H6 |
C1 |
H7 |
109.769 |
|
H6 |
C1 |
H8 |
109.783 |
| H7 |
C1 |
H8 |
108.149 |
|
H9 |
C4 |
H10 |
109.007 |
| H9 |
C4 |
H11 |
108.999 |
|
H10 |
C4 |
H11 |
107.783 |
| H12 |
C3 |
H13 |
106.818 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability