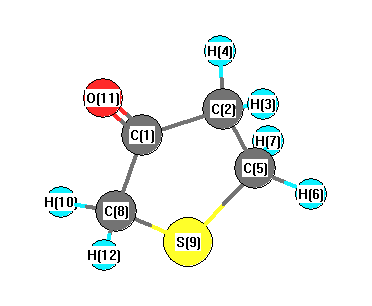
Vibrational Frequencies calculated at LSDA/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3086 |
3029 |
1.59 |
|
|
|
| 2 |
A |
3072 |
3014 |
0.94 |
|
|
|
| 3 |
A |
3066 |
3009 |
3.37 |
|
|
|
| 4 |
A |
3007 |
2951 |
19.57 |
|
|
|
| 5 |
A |
2987 |
2931 |
12.02 |
|
|
|
| 6 |
A |
2985 |
2929 |
1.62 |
|
|
|
| 7 |
A |
1834 |
1800 |
188.44 |
|
|
|
| 8 |
A |
1448 |
1421 |
5.36 |
|
|
|
| 9 |
A |
1395 |
1369 |
9.97 |
|
|
|
| 10 |
A |
1391 |
1365 |
17.61 |
|
|
|
| 11 |
A |
1265 |
1241 |
22.38 |
|
|
|
| 12 |
A |
1255 |
1232 |
14.54 |
|
|
|
| 13 |
A |
1200 |
1177 |
17.48 |
|
|
|
| 14 |
A |
1182 |
1160 |
29.60 |
|
|
|
| 15 |
A |
1139 |
1117 |
38.86 |
|
|
|
| 16 |
A |
1125 |
1104 |
4.28 |
|
|
|
| 17 |
A |
1063 |
1043 |
4.48 |
|
|
|
| 18 |
A |
999 |
980 |
12.08 |
|
|
|
| 19 |
A |
948 |
930 |
0.89 |
|
|
|
| 20 |
A |
854 |
838 |
9.95 |
|
|
|
| 21 |
A |
818 |
803 |
2.91 |
|
|
|
| 22 |
A |
793 |
778 |
0.59 |
|
|
|
| 23 |
A |
731 |
717 |
5.27 |
|
|
|
| 24 |
A |
677 |
664 |
0.53 |
|
|
|
| 25 |
A |
550 |
540 |
5.18 |
|
|
|
| 26 |
A |
476 |
467 |
3.95 |
|
|
|
| 27 |
A |
438 |
430 |
4.19 |
|
|
|
| 28 |
A |
416 |
408 |
4.40 |
|
|
|
| 29 |
A |
194 |
191 |
3.17 |
|
|
|
| 30 |
A |
71 |
69 |
12.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20231.6 cm
-1
Scaled (by 0.9813) Zero Point Vibrational Energy (zpe) 19853.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.397 |
|
|
|
| 2 |
C |
-0.415 |
|
|
|
| 3 |
H |
0.213 |
|
|
|
| 4 |
H |
0.228 |
|
|
|
| 5 |
C |
-0.500 |
|
|
|
| 6 |
H |
0.212 |
|
|
|
| 7 |
H |
0.217 |
|
|
|
| 8 |
C |
-0.570 |
|
|
|
| 9 |
S |
0.112 |
|
|
|
| 10 |
H |
0.241 |
|
|
|
| 11 |
O |
-0.371 |
|
|
|
| 12 |
H |
0.235 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.923 |
1.289 |
0.403 |
1.636 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-52.780 |
1.247 |
0.416 |
| y |
1.247 |
-37.819 |
-0.119 |
| z |
0.416 |
-0.119 |
-41.605 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-13.068 |
1.247 |
0.416 |
| y |
1.247 |
9.374 |
-0.119 |
| z |
0.416 |
-0.119 |
3.694 |
|
| Polar |
|---|
| 3z2-r2 | 7.388 |
|---|
| x2-y2 | -14.962 |
|---|
| xy | 1.247 |
|---|
| xz | 0.416 |
|---|
| yz | -0.119 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.926 |
0.085 |
-0.219 |
| y |
0.085 |
9.108 |
0.014 |
| z |
-0.219 |
0.014 |
6.470 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |