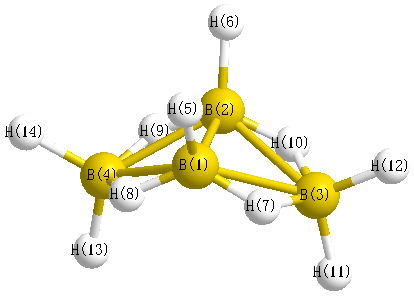
Vibrational Frequencies calculated at LSDA/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2641 |
2611 |
41.04 |
|
|
|
| 2 |
A1 |
2603 |
2573 |
23.37 |
|
|
|
| 3 |
A1 |
2504 |
2476 |
41.02 |
|
|
|
| 4 |
A1 |
2152 |
2127 |
0.17 |
|
|
|
| 5 |
A1 |
1559 |
1541 |
1.35 |
|
|
|
| 6 |
A1 |
1105 |
1092 |
2.25 |
|
|
|
| 7 |
A1 |
940 |
929 |
3.16 |
|
|
|
| 8 |
A1 |
828 |
819 |
8.94 |
|
|
|
| 9 |
A1 |
786 |
777 |
0.11 |
|
|
|
| 10 |
A1 |
638 |
630 |
1.48 |
|
|
|
| 11 |
A1 |
613 |
606 |
3.76 |
|
|
|
| 12 |
A1 |
205 |
203 |
11.13 |
|
|
|
| 13 |
A2 |
2204 |
2179 |
0.00 |
|
|
|
| 14 |
A2 |
1491 |
1474 |
0.00 |
|
|
|
| 15 |
A2 |
999 |
988 |
0.00 |
|
|
|
| 16 |
A2 |
952 |
941 |
0.00 |
|
|
|
| 17 |
A2 |
879 |
869 |
0.00 |
|
|
|
| 18 |
A2 |
716 |
708 |
0.00 |
|
|
|
| 19 |
A2 |
403 |
398 |
0.00 |
|
|
|
| 20 |
B1 |
2633 |
2604 |
21.25 |
|
|
|
| 21 |
B1 |
2176 |
2151 |
13.38 |
|
|
|
| 22 |
B1 |
1508 |
1490 |
10.04 |
|
|
|
| 23 |
B1 |
1000 |
989 |
11.91 |
|
|
|
| 24 |
B1 |
927 |
917 |
16.37 |
|
|
|
| 25 |
B1 |
865 |
856 |
16.15 |
|
|
|
| 26 |
B1 |
691 |
683 |
6.36 |
|
|
|
| 27 |
B1 |
592 |
586 |
4.33 |
|
|
|
| 28 |
B2 |
2606 |
2577 |
69.93 |
|
|
|
| 29 |
B2 |
2504 |
2475 |
47.45 |
|
|
|
| 30 |
B2 |
2179 |
2154 |
33.29 |
|
|
|
| 31 |
B2 |
1331 |
1316 |
8.03 |
|
|
|
| 32 |
B2 |
1088 |
1076 |
11.32 |
|
|
|
| 33 |
B2 |
879 |
869 |
17.28 |
|
|
|
| 34 |
B2 |
826 |
817 |
0.00 |
|
|
|
| 35 |
B2 |
631 |
623 |
5.53 |
|
|
|
| 36 |
B2 |
323 |
319 |
2.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23486.6 cm
-1
Scaled (by 0.9887) Zero Point Vibrational Energy (zpe) 23221.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
B |
-2.132 |
|
|
|
| 2 |
B |
-2.132 |
|
|
|
| 3 |
B |
-3.384 |
|
|
|
| 4 |
B |
-3.384 |
|
|
|
| 5 |
H |
0.968 |
|
|
|
| 6 |
H |
0.968 |
|
|
|
| 7 |
H |
1.045 |
|
|
|
| 8 |
H |
1.045 |
|
|
|
| 9 |
H |
1.045 |
|
|
|
| 10 |
H |
1.045 |
|
|
|
| 11 |
H |
1.294 |
|
|
|
| 12 |
H |
1.163 |
|
|
|
| 13 |
H |
1.294 |
|
|
|
| 14 |
H |
1.163 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.514 |
0.514 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.144 |
0.000 |
0.000 |
| y |
0.000 |
-34.423 |
0.000 |
| z |
0.000 |
0.000 |
-33.538 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.837 |
0.000 |
0.000 |
| y |
0.000 |
-3.082 |
0.000 |
| z |
0.000 |
0.000 |
-1.755 |
|
| Polar |
|---|
| 3z2-r2 | -3.510 |
|---|
| x2-y2 | 5.279 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.329 |
0.000 |
0.000 |
| y |
0.000 |
10.811 |
0.000 |
| z |
0.000 |
0.000 |
9.320 |
<r2> (average value of r
2) Å
2
| <r2> |
89.514 |
| (<r2>)1/2 |
9.461 |