Jump to
S1C2
Vibrational Frequencies calculated at LSDA/3-21G
Geometric Data calculated at LSDA/3-21G
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at LSDA/3-21G
| | hartrees |
|---|
| Energy at 0K | -233.261948 |
| Energy at 298.15K | -233.275360 |
| HF Energy | -233.261948 |
| Nuclear repulsion energy | 250.390038 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3077 |
3026 |
33.79 |
|
|
|
| 2 |
A' |
3057 |
3007 |
32.71 |
|
|
|
| 3 |
A' |
3049 |
2999 |
18.64 |
|
|
|
| 4 |
A' |
3029 |
2979 |
8.13 |
|
|
|
| 5 |
A' |
3012 |
2962 |
8.14 |
|
|
|
| 6 |
A' |
3001 |
2952 |
1.33 |
|
|
|
| 7 |
A' |
2973 |
2924 |
17.18 |
|
|
|
| 8 |
A' |
2969 |
2920 |
18.43 |
|
|
|
| 9 |
A' |
1497 |
1473 |
18.34 |
|
|
|
| 10 |
A' |
1492 |
1467 |
7.26 |
|
|
|
| 11 |
A' |
1465 |
1441 |
1.31 |
|
|
|
| 12 |
A' |
1398 |
1375 |
27.54 |
|
|
|
| 13 |
A' |
1391 |
1369 |
6.67 |
|
|
|
| 14 |
A' |
1349 |
1327 |
0.05 |
|
|
|
| 15 |
A' |
1319 |
1298 |
3.77 |
|
|
|
| 16 |
A' |
1254 |
1234 |
11.28 |
|
|
|
| 17 |
A' |
1223 |
1203 |
11.82 |
|
|
|
| 18 |
A' |
1191 |
1171 |
2.05 |
|
|
|
| 19 |
A' |
1134 |
1115 |
0.16 |
|
|
|
| 20 |
A' |
1043 |
1026 |
0.28 |
|
|
|
| 21 |
A' |
964 |
948 |
3.02 |
|
|
|
| 22 |
A' |
948 |
933 |
4.15 |
|
|
|
| 23 |
A' |
864 |
850 |
3.08 |
|
|
|
| 24 |
A' |
811 |
798 |
2.77 |
|
|
|
| 25 |
A' |
657 |
646 |
3.20 |
|
|
|
| 26 |
A' |
424 |
417 |
0.31 |
|
|
|
| 27 |
A' |
325 |
320 |
0.21 |
|
|
|
| 28 |
A' |
126 |
124 |
0.03 |
|
|
|
| 29 |
A" |
3072 |
3022 |
10.36 |
|
|
|
| 30 |
A" |
3055 |
3005 |
15.37 |
|
|
|
| 31 |
A" |
3053 |
3003 |
13.37 |
|
|
|
| 32 |
A" |
2999 |
2950 |
32.28 |
|
|
|
| 33 |
A" |
1495 |
1471 |
4.94 |
|
|
|
| 34 |
A" |
1494 |
1470 |
4.52 |
|
|
|
| 35 |
A" |
1443 |
1419 |
14.37 |
|
|
|
| 36 |
A" |
1250 |
1229 |
0.10 |
|
|
|
| 37 |
A" |
1246 |
1226 |
0.01 |
|
|
|
| 38 |
A" |
1195 |
1176 |
0.31 |
|
|
|
| 39 |
A" |
1118 |
1099 |
0.00 |
|
|
|
| 40 |
A" |
1114 |
1095 |
1.91 |
|
|
|
| 41 |
A" |
995 |
978 |
10.51 |
|
|
|
| 42 |
A" |
900 |
885 |
0.04 |
|
|
|
| 43 |
A" |
882 |
867 |
0.04 |
|
|
|
| 44 |
A" |
793 |
780 |
0.59 |
|
|
|
| 45 |
A" |
374 |
368 |
0.01 |
|
|
|
| 46 |
A" |
276 |
272 |
0.45 |
|
|
|
| 47 |
A" |
262 |
258 |
0.05 |
|
|
|
| 48 |
A" |
231 |
227 |
0.28 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36143.1 cm
-1
Scaled (by 0.9836) Zero Point Vibrational Energy (zpe) 35550.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.255 |
0.084 |
1.087 |
| C2 |
0.255 |
0.084 |
-1.087 |
| C3 |
-0.672 |
0.697 |
0.000 |
| C4 |
0.869 |
-0.847 |
0.000 |
| C5 |
-0.862 |
2.194 |
0.000 |
| C6 |
0.255 |
-2.229 |
0.000 |
| H7 |
0.987 |
0.828 |
1.451 |
| H8 |
-0.230 |
-0.408 |
1.947 |
| H9 |
0.987 |
0.828 |
-1.451 |
| H10 |
-0.230 |
-0.408 |
-1.947 |
| H11 |
-1.650 |
0.181 |
0.000 |
| H12 |
1.970 |
-0.909 |
0.000 |
| H13 |
0.122 |
2.699 |
0.000 |
| H14 |
-0.849 |
-2.159 |
0.000 |
| H15 |
-1.420 |
2.522 |
-0.896 |
| H16 |
-1.420 |
2.522 |
0.896 |
| H17 |
0.565 |
-2.795 |
-0.897 |
| H18 |
0.565 |
-2.795 |
0.897 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.1747 | 1.5553 | 1.5575 | 2.6233 | 2.5562 | 1.1047 | 1.1030 | 2.7444 | 3.1121 | 2.1960 | 2.2604 | 2.8347 | 2.7259 | 3.5610 | 2.9639 | 3.5105 | 2.9022 |
C2 | 2.1747 | | 1.5553 | 1.5575 | 2.6233 | 2.5562 | 2.7444 | 3.1121 | 1.1047 | 1.1030 | 2.1960 | 2.2604 | 2.8347 | 2.7259 | 2.9639 | 3.5610 | 2.9022 | 3.5105 | C3 | 1.5553 | 1.5553 | | 2.1817 | 1.5089 | 3.0699 | 2.2082 | 2.2821 | 2.2082 | 2.2821 | 1.1056 | 3.0924 | 2.1535 | 2.8611 | 2.1656 | 2.1656 | 3.8122 | 3.8122 | C4 | 1.5575 | 1.5575 | 2.1817 | | 3.4990 | 1.5121 | 2.2192 | 2.2784 | 2.2192 | 2.2784 | 2.7205 | 1.1035 | 3.6234 | 2.1607 | 4.1699 | 4.1699 | 2.1659 | 2.1659 | C5 | 2.6233 | 2.6233 | 1.5089 | 3.4990 | | 4.5622 | 2.7183 | 3.3106 | 2.7183 | 3.3106 | 2.1621 | 4.2008 | 1.1058 | 4.3527 | 1.1051 | 1.1051 | 5.2663 | 5.2663 | C6 | 2.5562 | 2.5562 | 3.0699 | 1.5121 | 4.5622 | | 3.4621 | 2.7100 | 3.4621 | 2.7100 | 3.0724 | 2.1646 | 4.9297 | 1.1061 | 5.1166 | 5.1166 | 1.1050 | 1.1050 | H7 | 1.1047 | 2.7444 | 2.2082 | 2.2192 | 2.7183 | 3.4621 | | 1.8037 | 2.9022 | 3.8151 | 3.0786 | 2.4673 | 2.5205 | 3.7937 | 3.7642 | 2.9950 | 4.3378 | 3.6892 | H8 | 1.1030 | 3.1121 | 2.2821 | 2.2784 | 3.3106 | 2.7100 | 1.8037 | | 3.8151 | 3.8942 | 2.4808 | 2.9805 | 3.6831 | 2.6905 | 4.2520 | 3.3321 | 3.7975 | 2.7267 | H9 | 2.7444 | 1.1047 | 2.2082 | 2.2192 | 2.7183 | 3.4621 | 2.9022 | 3.8151 | | 1.8037 | 3.0786 | 2.4673 | 2.5205 | 3.7937 | 2.9950 | 3.7642 | 3.6892 | 4.3378 | H10 | 3.1121 | 1.1030 | 2.2821 | 2.2784 | 3.3106 | 2.7100 | 3.8151 | 3.8942 | 1.8037 | | 2.4808 | 2.9805 | 3.6831 | 2.6905 | 3.3321 | 4.2520 | 2.7267 | 3.7975 | H11 | 2.1960 | 2.1960 | 1.1056 | 2.7205 | 2.1621 | 3.0724 | 3.0786 | 2.4808 | 3.0786 | 2.4808 | | 3.7809 | 3.0790 | 2.4730 | 2.5168 | 2.5168 | 3.8171 | 3.8171 | H12 | 2.2604 | 2.2604 | 3.0924 | 1.1035 | 4.2008 | 2.1646 | 2.4673 | 2.9805 | 2.4673 | 2.9805 | 3.7809 | | 4.0530 | 3.0836 | 4.9054 | 4.9054 | 2.5174 | 2.5174 | H13 | 2.8347 | 2.8347 | 2.1535 | 3.6234 | 1.1058 | 4.9297 | 2.5205 | 3.6831 | 2.5205 | 3.6831 | 3.0790 | 4.0530 | | 4.9533 | 1.7923 | 1.7923 | 5.5841 | 5.5841 | H14 | 2.7259 | 2.7259 | 2.8611 | 2.1607 | 4.3527 | 1.1061 | 3.7937 | 2.6905 | 3.7937 | 2.6905 | 2.4730 | 3.0836 | 4.9533 | | 4.7993 | 4.7993 | 1.7914 | 1.7914 | H15 | 3.5610 | 2.9639 | 2.1656 | 4.1699 | 1.1051 | 5.1166 | 3.7642 | 4.2520 | 2.9950 | 3.3321 | 2.5168 | 4.9054 | 1.7923 | 4.7993 | | 1.7915 | 5.6754 | 5.9518 | H16 | 2.9639 | 3.5610 | 2.1656 | 4.1699 | 1.1051 | 5.1166 | 2.9950 | 3.3321 | 3.7642 | 4.2520 | 2.5168 | 4.9054 | 1.7923 | 4.7993 | 1.7915 | | 5.9518 | 5.6754 | H17 | 3.5105 | 2.9022 | 3.8122 | 2.1659 | 5.2663 | 1.1050 | 4.3378 | 3.7975 | 3.6892 | 2.7267 | 3.8171 | 2.5174 | 5.5841 | 1.7914 | 5.6754 | 5.9518 | | 1.7939 | H18 | 2.9022 | 3.5105 | 3.8122 | 2.1659 | 5.2663 | 1.1050 | 3.6892 | 2.7267 | 4.3378 | 3.7975 | 3.8171 | 2.5174 | 5.5841 | 1.7914 | 5.9518 | 5.6754 | 1.7939 | |
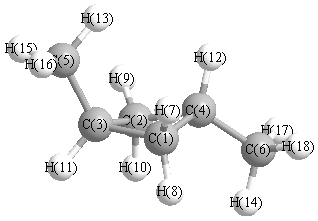 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
88.717 |
|
C1 |
C3 |
C5 |
117.765 |
| C1 |
C3 |
H11 |
110.087 |
|
C1 |
C4 |
C2 |
88.558 |
| C1 |
C4 |
C6 |
112.755 |
|
C1 |
C4 |
H12 |
115.245 |
| C2 |
C3 |
C5 |
117.765 |
|
C2 |
C3 |
H11 |
110.087 |
| C2 |
C4 |
C6 |
112.755 |
|
C2 |
C4 |
H12 |
115.245 |
| C3 |
C1 |
C4 |
88.996 |
|
C3 |
C1 |
H7 |
111.092 |
| C3 |
C1 |
H8 |
117.279 |
|
C3 |
C2 |
C4 |
88.996 |
| C3 |
C2 |
H9 |
111.092 |
|
C3 |
C2 |
H10 |
117.279 |
| C3 |
C5 |
H13 |
109.943 |
|
C3 |
C5 |
H15 |
110.940 |
| C3 |
C5 |
H16 |
110.940 |
|
C4 |
C1 |
H7 |
111.811 |
| C4 |
C1 |
H8 |
116.793 |
|
C4 |
C2 |
H9 |
111.811 |
| C4 |
C2 |
H10 |
116.793 |
|
C4 |
C6 |
H14 |
110.264 |
| C4 |
C6 |
H17 |
110.749 |
|
C4 |
C6 |
H18 |
110.749 |
| C5 |
C3 |
H11 |
110.634 |
|
C6 |
C4 |
H12 |
110.736 |
| H7 |
C1 |
H8 |
109.566 |
|
H9 |
C2 |
H10 |
109.566 |
| H13 |
C5 |
H15 |
108.318 |
|
H13 |
C5 |
H16 |
108.318 |
| H14 |
C6 |
H17 |
108.231 |
|
H14 |
C6 |
H18 |
108.231 |
| H15 |
C5 |
H16 |
108.297 |
|
H17 |
C6 |
H18 |
108.534 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.368 |
|
|
|
| 2 |
C |
-0.368 |
|
|
|
| 3 |
C |
-0.243 |
|
|
|
| 4 |
C |
-0.277 |
|
|
|
| 5 |
C |
-0.623 |
|
|
|
| 6 |
C |
-0.602 |
|
|
|
| 7 |
H |
0.208 |
|
|
|
| 8 |
H |
0.199 |
|
|
|
| 9 |
H |
0.208 |
|
|
|
| 10 |
H |
0.199 |
|
|
|
| 11 |
H |
0.210 |
|
|
|
| 12 |
H |
0.199 |
|
|
|
| 13 |
H |
0.203 |
|
|
|
| 14 |
H |
0.208 |
|
|
|
| 15 |
H |
0.213 |
|
|
|
| 16 |
H |
0.213 |
|
|
|
| 17 |
H |
0.211 |
|
|
|
| 18 |
H |
0.211 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.065 |
0.038 |
0.000 |
0.075 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.381 |
0.353 |
0.000 |
| y |
0.353 |
-39.737 |
0.000 |
| z |
0.000 |
0.000 |
-39.930 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.453 |
0.353 |
0.000 |
| y |
0.353 |
-0.082 |
0.000 |
| z |
0.000 |
0.000 |
-0.371 |
|
| Polar |
|---|
| 3z2-r2 | -0.742 |
|---|
| x2-y2 | 0.356 |
|---|
| xy | 0.353 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.210 |
-0.579 |
0.000 |
| y |
-0.579 |
9.686 |
0.000 |
| z |
0.000 |
0.000 |
8.446 |
<r2> (average value of r
2) Å
2
| <r2> |
187.605 |
| (<r2>)1/2 |
13.697 |