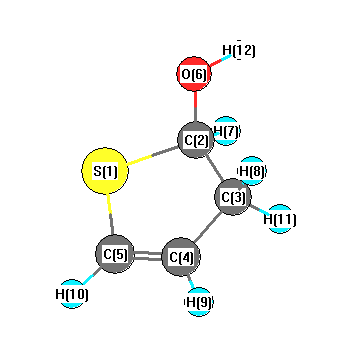
Vibrational Frequencies calculated at LSDA/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3750 |
3704 |
68.01 |
|
|
|
| 2 |
A |
3151 |
3112 |
2.49 |
|
|
|
| 3 |
A |
3130 |
3091 |
0.29 |
|
|
|
| 4 |
A |
3022 |
2984 |
9.28 |
|
|
|
| 5 |
A |
2927 |
2891 |
38.60 |
|
|
|
| 6 |
A |
2910 |
2874 |
10.00 |
|
|
|
| 7 |
A |
1614 |
1594 |
15.77 |
|
|
|
| 8 |
A |
1400 |
1382 |
8.75 |
|
|
|
| 9 |
A |
1383 |
1366 |
3.24 |
|
|
|
| 10 |
A |
1295 |
1280 |
11.50 |
|
|
|
| 11 |
A |
1256 |
1240 |
50.30 |
|
|
|
| 12 |
A |
1231 |
1216 |
7.91 |
|
|
|
| 13 |
A |
1211 |
1196 |
47.60 |
|
|
|
| 14 |
A |
1142 |
1128 |
175.01 |
|
|
|
| 15 |
A |
1103 |
1089 |
24.07 |
|
|
|
| 16 |
A |
1076 |
1062 |
9.16 |
|
|
|
| 17 |
A |
1013 |
1000 |
4.31 |
|
|
|
| 18 |
A |
972 |
960 |
14.49 |
|
|
|
| 19 |
A |
929 |
917 |
10.00 |
|
|
|
| 20 |
A |
879 |
868 |
1.18 |
|
|
|
| 21 |
A |
797 |
787 |
34.56 |
|
|
|
| 22 |
A |
685 |
676 |
9.19 |
|
|
|
| 23 |
A |
638 |
630 |
52.03 |
|
|
|
| 24 |
A |
616 |
609 |
46.23 |
|
|
|
| 25 |
A |
481 |
475 |
4.16 |
|
|
|
| 26 |
A |
450 |
444 |
5.18 |
|
|
|
| 27 |
A |
399 |
394 |
1.84 |
|
|
|
| 28 |
A |
360 |
356 |
117.51 |
|
|
|
| 29 |
A |
313 |
309 |
13.12 |
|
|
|
| 30 |
A |
97 |
96 |
1.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20113.6 cm
-1
Scaled (by 0.9877) Zero Point Vibrational Energy (zpe) 19866.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.143 |
|
|
|
| 2 |
C |
-0.218 |
|
|
|
| 3 |
C |
-0.290 |
|
|
|
| 4 |
C |
-0.147 |
|
|
|
| 5 |
C |
-0.298 |
|
|
|
| 6 |
O |
-0.321 |
|
|
|
| 7 |
H |
0.193 |
|
|
|
| 8 |
H |
0.191 |
|
|
|
| 9 |
H |
0.139 |
|
|
|
| 10 |
H |
0.168 |
|
|
|
| 11 |
H |
0.172 |
|
|
|
| 12 |
H |
0.268 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.449 |
0.718 |
1.439 |
1.669 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.594 |
-2.936 |
3.267 |
| y |
-2.936 |
-43.008 |
-0.387 |
| z |
3.267 |
-0.387 |
-45.496 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.658 |
-2.936 |
3.267 |
| y |
-2.936 |
-1.463 |
-0.387 |
| z |
3.267 |
-0.387 |
-5.194 |
|
| Polar |
|---|
| 3z2-r2 | -10.389 |
|---|
| x2-y2 | 5.414 |
|---|
| xy | -2.936 |
|---|
| xz | 3.267 |
|---|
| yz | -0.387 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.566 |
-0.076 |
0.119 |
| y |
-0.076 |
11.624 |
0.108 |
| z |
0.119 |
0.108 |
6.275 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |