Jump to
S1C2
Energy calculated at LSDA/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -310.605084 |
| Energy at 298.15K | -310.620422 |
| HF Energy | -310.605084 |
| Nuclear repulsion energy | 316.705375 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3072 |
3039 |
18.18 |
|
|
|
| 2 |
A1 |
2978 |
2946 |
47.17 |
|
|
|
| 3 |
A1 |
2970 |
2938 |
7.60 |
|
|
|
| 4 |
A1 |
2847 |
2815 |
94.82 |
|
|
|
| 5 |
A1 |
1443 |
1427 |
4.33 |
|
|
|
| 6 |
A1 |
1415 |
1399 |
6.84 |
|
|
|
| 7 |
A1 |
1396 |
1381 |
0.02 |
|
|
|
| 8 |
A1 |
1384 |
1369 |
0.38 |
|
|
|
| 9 |
A1 |
1329 |
1315 |
1.65 |
|
|
|
| 10 |
A1 |
1259 |
1245 |
4.55 |
|
|
|
| 11 |
A1 |
1156 |
1143 |
4.22 |
|
|
|
| 12 |
A1 |
1075 |
1063 |
10.59 |
|
|
|
| 13 |
A1 |
1042 |
1031 |
15.77 |
|
|
|
| 14 |
A1 |
894 |
884 |
11.48 |
|
|
|
| 15 |
A1 |
421 |
417 |
0.30 |
|
|
|
| 16 |
A1 |
320 |
316 |
0.13 |
|
|
|
| 17 |
A1 |
97 |
96 |
0.12 |
|
|
|
| 18 |
A2 |
3052 |
3019 |
0.00 |
|
|
|
| 19 |
A2 |
3022 |
2989 |
0.00 |
|
|
|
| 20 |
A2 |
2872 |
2840 |
0.00 |
|
|
|
| 21 |
A2 |
1395 |
1380 |
0.00 |
|
|
|
| 22 |
A2 |
1239 |
1225 |
0.00 |
|
|
|
| 23 |
A2 |
1193 |
1180 |
0.00 |
|
|
|
| 24 |
A2 |
1120 |
1107 |
0.00 |
|
|
|
| 25 |
A2 |
851 |
841 |
0.00 |
|
|
|
| 26 |
A2 |
744 |
736 |
0.00 |
|
|
|
| 27 |
A2 |
233 |
230 |
0.00 |
|
|
|
| 28 |
A2 |
141 |
140 |
0.00 |
|
|
|
| 29 |
A2 |
78 |
77 |
0.00 |
|
|
|
| 30 |
B1 |
3052 |
3019 |
58.16 |
|
|
|
| 31 |
B1 |
3022 |
2989 |
11.00 |
|
|
|
| 32 |
B1 |
2861 |
2829 |
128.35 |
|
|
|
| 33 |
B1 |
1395 |
1380 |
21.04 |
|
|
|
| 34 |
B1 |
1236 |
1222 |
0.45 |
|
|
|
| 35 |
B1 |
1198 |
1185 |
5.67 |
|
|
|
| 36 |
B1 |
1143 |
1131 |
3.08 |
|
|
|
| 37 |
B1 |
861 |
851 |
4.18 |
|
|
|
| 38 |
B1 |
743 |
735 |
7.94 |
|
|
|
| 39 |
B1 |
233 |
231 |
0.23 |
|
|
|
| 40 |
B1 |
156 |
154 |
3.66 |
|
|
|
| 41 |
B1 |
56 |
56 |
0.17 |
|
|
|
| 42 |
B2 |
3072 |
3038 |
12.22 |
|
|
|
| 43 |
B2 |
2978 |
2945 |
4.44 |
|
|
|
| 44 |
B2 |
2970 |
2938 |
24.12 |
|
|
|
| 45 |
B2 |
2836 |
2805 |
19.36 |
|
|
|
| 46 |
B2 |
1419 |
1404 |
16.91 |
|
|
|
| 47 |
B2 |
1409 |
1393 |
0.94 |
|
|
|
| 48 |
B2 |
1394 |
1379 |
2.86 |
|
|
|
| 49 |
B2 |
1343 |
1329 |
17.00 |
|
|
|
| 50 |
B2 |
1323 |
1308 |
35.82 |
|
|
|
| 51 |
B2 |
1244 |
1230 |
2.83 |
|
|
|
| 52 |
B2 |
1213 |
1200 |
233.03 |
|
|
|
| 53 |
B2 |
1104 |
1092 |
3.32 |
|
|
|
| 54 |
B2 |
1068 |
1056 |
0.02 |
|
|
|
| 55 |
B2 |
884 |
874 |
3.05 |
|
|
|
| 56 |
B2 |
511 |
505 |
6.69 |
|
|
|
| 57 |
B2 |
235 |
233 |
0.47 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40997.9 cm
-1
Scaled (by 0.989) Zero Point Vibrational Energy (zpe) 40546.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/cc-pVDZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.416 |
| C2 |
0.000 |
1.156 |
-0.363 |
| C3 |
0.000 |
-1.156 |
-0.363 |
| C4 |
0.000 |
2.363 |
0.531 |
| C5 |
0.000 |
-2.363 |
0.531 |
| C6 |
0.000 |
3.649 |
-0.259 |
| C7 |
0.000 |
-3.649 |
-0.259 |
| H8 |
0.897 |
1.174 |
-1.041 |
| H9 |
-0.897 |
1.174 |
-1.041 |
| H10 |
-0.897 |
-1.174 |
-1.041 |
| H11 |
0.897 |
-1.174 |
-1.041 |
| H12 |
0.889 |
2.305 |
1.197 |
| H13 |
-0.889 |
2.305 |
1.197 |
| H14 |
-0.889 |
-2.305 |
1.197 |
| H15 |
0.889 |
-2.305 |
1.197 |
| H16 |
0.000 |
4.540 |
0.400 |
| H17 |
-0.894 |
3.721 |
-0.915 |
| H18 |
0.894 |
3.721 |
-0.915 |
| H19 |
0.000 |
-4.540 |
0.400 |
| H20 |
0.894 |
-3.721 |
-0.915 |
| H21 |
-0.894 |
-3.721 |
-0.915 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.3938 | 1.3938 | 2.3660 | 2.3660 | 3.7111 | 3.7111 | 2.0751 | 2.0751 | 2.0751 | 2.0751 | 2.5906 | 2.5906 | 2.5906 | 2.5906 | 4.5397 | 4.0524 | 4.0524 | 4.5397 | 4.0524 | 4.0524 |
C2 | 1.3938 | | 2.3116 | 1.5020 | 3.6307 | 2.4955 | 4.8061 | 1.1247 | 1.1247 | 2.5869 | 2.5869 | 2.1311 | 2.1311 | 3.8983 | 3.8983 | 3.4688 | 2.7726 | 2.7726 | 5.7464 | 4.9892 | 4.9892 | C3 | 1.3938 | 2.3116 | | 3.6307 | 1.5020 | 4.8061 | 2.4955 | 2.5869 | 2.5869 | 1.1247 | 1.1247 | 3.8983 | 3.8983 | 2.1311 | 2.1311 | 5.7464 | 4.9892 | 4.9892 | 3.4688 | 2.7726 | 2.7726 | C4 | 2.3660 | 1.5020 | 3.6307 | | 4.7265 | 1.5092 | 6.0641 | 2.1655 | 2.1655 | 3.9731 | 3.9731 | 1.1122 | 1.1122 | 4.7983 | 4.7983 | 2.1804 | 2.1761 | 2.1761 | 6.9042 | 6.3178 | 6.3178 | C5 | 2.3660 | 3.6307 | 1.5020 | 4.7265 | | 6.0641 | 1.5092 | 3.9731 | 3.9731 | 2.1655 | 2.1655 | 4.7983 | 4.7983 | 1.1122 | 1.1122 | 6.9042 | 6.3178 | 6.3178 | 2.1804 | 2.1761 | 2.1761 | C6 | 3.7111 | 2.4955 | 4.8061 | 1.5092 | 6.0641 | | 7.2983 | 2.7464 | 2.7464 | 4.9676 | 4.9676 | 2.1719 | 2.1719 | 6.1934 | 6.1934 | 1.1081 | 1.1112 | 1.1112 | 8.2154 | 7.4536 | 7.4536 | C7 | 3.7111 | 4.8061 | 2.4955 | 6.0641 | 1.5092 | 7.2983 | | 4.9676 | 4.9676 | 2.7464 | 2.7464 | 6.1934 | 6.1934 | 2.1719 | 2.1719 | 8.2154 | 7.4536 | 7.4536 | 1.1081 | 1.1112 | 1.1112 | H8 | 2.0751 | 1.1247 | 2.5869 | 2.1655 | 3.9731 | 2.7464 | 4.9676 | | 1.7939 | 2.9547 | 2.3477 | 2.5071 | 3.0781 | 4.5052 | 4.1361 | 3.7697 | 3.1166 | 2.5507 | 5.9604 | 4.8970 | 5.2141 | H9 | 2.0751 | 1.1247 | 2.5869 | 2.1655 | 3.9731 | 2.7464 | 4.9676 | 1.7939 | | 2.3477 | 2.9547 | 3.0781 | 2.5071 | 4.1361 | 4.5052 | 3.7697 | 2.5507 | 3.1166 | 5.9604 | 5.2141 | 4.8970 | H10 | 2.0751 | 2.5869 | 1.1247 | 3.9731 | 2.1655 | 4.9676 | 2.7464 | 2.9547 | 2.3477 | | 1.7939 | 4.5052 | 4.1361 | 2.5071 | 3.0781 | 5.9604 | 4.8970 | 5.2141 | 3.7697 | 3.1166 | 2.5507 | H11 | 2.0751 | 2.5869 | 1.1247 | 3.9731 | 2.1655 | 4.9676 | 2.7464 | 2.3477 | 2.9547 | 1.7939 | | 4.1361 | 4.5052 | 3.0781 | 2.5071 | 5.9604 | 5.2141 | 4.8970 | 3.7697 | 2.5507 | 3.1166 | H12 | 2.5906 | 2.1311 | 3.8983 | 1.1122 | 4.7983 | 2.1719 | 6.1934 | 2.5071 | 3.0781 | 4.5052 | 4.1361 | | 1.7778 | 4.9404 | 4.6095 | 2.5337 | 3.1059 | 2.5433 | 6.9477 | 6.3856 | 6.6298 | H13 | 2.5906 | 2.1311 | 3.8983 | 1.1122 | 4.7983 | 2.1719 | 6.1934 | 3.0781 | 2.5071 | 4.1361 | 4.5052 | 1.7778 | | 4.6095 | 4.9404 | 2.5337 | 2.5433 | 3.1059 | 6.9477 | 6.6298 | 6.3856 | H14 | 2.5906 | 3.8983 | 2.1311 | 4.7983 | 1.1122 | 6.1934 | 2.1719 | 4.5052 | 4.1361 | 2.5071 | 3.0781 | 4.9404 | 4.6095 | | 1.7778 | 6.9477 | 6.3856 | 6.6298 | 2.5337 | 3.1059 | 2.5433 | H15 | 2.5906 | 3.8983 | 2.1311 | 4.7983 | 1.1122 | 6.1934 | 2.1719 | 4.1361 | 4.5052 | 3.0781 | 2.5071 | 4.6095 | 4.9404 | 1.7778 | | 6.9477 | 6.6298 | 6.3856 | 2.5337 | 2.5433 | 3.1059 | H16 | 4.5397 | 3.4688 | 5.7464 | 2.1804 | 6.9042 | 1.1081 | 8.2154 | 3.7697 | 3.7697 | 5.9604 | 5.9604 | 2.5337 | 2.5337 | 6.9477 | 6.9477 | | 1.7887 | 1.7887 | 9.0794 | 8.4129 | 8.4129 | H17 | 4.0524 | 2.7726 | 4.9892 | 2.1761 | 6.3178 | 1.1112 | 7.4536 | 3.1166 | 2.5507 | 4.8970 | 5.2141 | 3.1059 | 2.5433 | 6.3856 | 6.6298 | 1.7887 | | 1.7876 | 8.4129 | 7.6546 | 7.4430 | H18 | 4.0524 | 2.7726 | 4.9892 | 2.1761 | 6.3178 | 1.1112 | 7.4536 | 2.5507 | 3.1166 | 5.2141 | 4.8970 | 2.5433 | 3.1059 | 6.6298 | 6.3856 | 1.7887 | 1.7876 | | 8.4129 | 7.4430 | 7.6546 | H19 | 4.5397 | 5.7464 | 3.4688 | 6.9042 | 2.1804 | 8.2154 | 1.1081 | 5.9604 | 5.9604 | 3.7697 | 3.7697 | 6.9477 | 6.9477 | 2.5337 | 2.5337 | 9.0794 | 8.4129 | 8.4129 | | 1.7887 | 1.7887 | H20 | 4.0524 | 4.9892 | 2.7726 | 6.3178 | 2.1761 | 7.4536 | 1.1112 | 4.8970 | 5.2141 | 3.1166 | 2.5507 | 6.3856 | 6.6298 | 3.1059 | 2.5433 | 8.4129 | 7.6546 | 7.4430 | 1.7887 | | 1.7876 | H21 | 4.0524 | 4.9892 | 2.7726 | 6.3178 | 2.1761 | 7.4536 | 1.1112 | 5.2141 | 4.8970 | 2.5507 | 3.1166 | 6.6298 | 6.3856 | 2.5433 | 3.1059 | 8.4129 | 7.4430 | 7.6546 | 1.7887 | 1.7876 | |
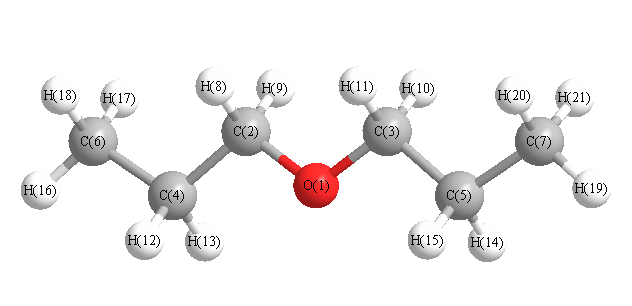 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
109.524 |
|
O1 |
C2 |
H8 |
110.509 |
| O1 |
C2 |
H9 |
110.509 |
|
O1 |
C3 |
C5 |
109.524 |
| O1 |
C3 |
H10 |
110.509 |
|
O1 |
C3 |
H11 |
110.509 |
| C2 |
O1 |
C3 |
112.046 |
|
C2 |
C4 |
C6 |
111.940 |
| C2 |
C4 |
H12 |
108.291 |
|
C2 |
C4 |
H13 |
108.291 |
| C3 |
C5 |
C7 |
111.940 |
|
C3 |
C5 |
H14 |
108.291 |
| C3 |
C5 |
H15 |
108.291 |
|
C4 |
C2 |
H8 |
110.230 |
| C4 |
C2 |
H9 |
110.230 |
|
C4 |
C6 |
H16 |
111.923 |
| C4 |
C6 |
H17 |
111.382 |
|
C4 |
C6 |
H18 |
111.382 |
| C5 |
C3 |
H10 |
110.230 |
|
C5 |
C3 |
H11 |
110.230 |
| C5 |
C7 |
H19 |
111.923 |
|
C5 |
C7 |
H20 |
111.382 |
| C5 |
C7 |
H21 |
111.382 |
|
C6 |
C4 |
H12 |
110.989 |
| C6 |
C4 |
H13 |
110.989 |
|
C7 |
C5 |
H14 |
110.989 |
| C7 |
C5 |
H15 |
110.989 |
|
H8 |
C2 |
H9 |
105.789 |
| H10 |
C3 |
H11 |
105.789 |
|
H12 |
C4 |
H13 |
106.113 |
| H14 |
C5 |
H15 |
106.113 |
|
H16 |
C6 |
H17 |
107.405 |
| H16 |
C6 |
H18 |
107.405 |
|
H17 |
C6 |
H18 |
107.093 |
| H19 |
C7 |
H20 |
107.405 |
|
H19 |
C7 |
H21 |
107.405 |
| H20 |
C7 |
H21 |
107.093 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.224 |
|
|
|
| 2 |
C |
0.026 |
|
|
|
| 3 |
C |
0.026 |
|
|
|
| 4 |
C |
-0.135 |
|
|
|
| 5 |
C |
-0.135 |
|
|
|
| 6 |
C |
-0.171 |
|
|
|
| 7 |
C |
-0.171 |
|
|
|
| 8 |
H |
0.032 |
|
|
|
| 9 |
H |
0.032 |
|
|
|
| 10 |
H |
0.032 |
|
|
|
| 11 |
H |
0.032 |
|
|
|
| 12 |
H |
0.064 |
|
|
|
| 13 |
H |
0.064 |
|
|
|
| 14 |
H |
0.064 |
|
|
|
| 15 |
H |
0.064 |
|
|
|
| 16 |
H |
0.067 |
|
|
|
| 17 |
H |
0.066 |
|
|
|
| 18 |
H |
0.066 |
|
|
|
| 19 |
H |
0.067 |
|
|
|
| 20 |
H |
0.066 |
|
|
|
| 21 |
H |
0.066 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.691 |
0.691 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.436 |
0.000 |
0.000 |
| y |
0.000 |
-43.710 |
0.000 |
| z |
0.000 |
0.000 |
-47.491 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.835 |
0.000 |
0.000 |
| y |
0.000 |
3.253 |
0.000 |
| z |
0.000 |
0.000 |
-2.418 |
|
| Polar |
|---|
| 3z2-r2 | -4.836 |
|---|
| x2-y2 | -2.725 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.953 |
0.000 |
0.000 |
| y |
0.000 |
14.264 |
0.000 |
| z |
0.000 |
0.000 |
10.381 |
<r2> (average value of r
2) Å
2
| <r2> |
425.335 |
| (<r2>)1/2 |
20.624 |
Jump to
S1C1
Energy calculated at LSDA/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -310.607661 |
| Energy at 298.15K | -310.623271 |
| HF Energy | -310.607661 |
| Nuclear repulsion energy | 328.434319 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3081 |
3048 |
0.22 |
|
|
|
| 2 |
A |
3062 |
3028 |
33.25 |
|
|
|
| 3 |
A |
3024 |
2991 |
9.97 |
|
|
|
| 4 |
A |
2977 |
2944 |
2.07 |
|
|
|
| 5 |
A |
2974 |
2942 |
3.16 |
|
|
|
| 6 |
A |
2896 |
2864 |
13.50 |
|
|
|
| 7 |
A |
2854 |
2823 |
88.67 |
|
|
|
| 8 |
A |
1433 |
1417 |
7.88 |
|
|
|
| 9 |
A |
1415 |
1399 |
4.10 |
|
|
|
| 10 |
A |
1389 |
1373 |
8.21 |
|
|
|
| 11 |
A |
1375 |
1360 |
3.80 |
|
|
|
| 12 |
A |
1372 |
1357 |
0.36 |
|
|
|
| 13 |
A |
1332 |
1318 |
4.69 |
|
|
|
| 14 |
A |
1290 |
1276 |
1.08 |
|
|
|
| 15 |
A |
1251 |
1237 |
5.08 |
|
|
|
| 16 |
A |
1203 |
1190 |
1.33 |
|
|
|
| 17 |
A |
1136 |
1124 |
14.12 |
|
|
|
| 18 |
A |
1105 |
1093 |
8.79 |
|
|
|
| 19 |
A |
1092 |
1080 |
8.62 |
|
|
|
| 20 |
A |
936 |
926 |
3.51 |
|
|
|
| 21 |
A |
914 |
904 |
2.97 |
|
|
|
| 22 |
A |
877 |
867 |
4.86 |
|
|
|
| 23 |
A |
726 |
718 |
0.10 |
|
|
|
| 24 |
A |
475 |
470 |
0.26 |
|
|
|
| 25 |
A |
334 |
331 |
0.42 |
|
|
|
| 26 |
A |
269 |
266 |
0.92 |
|
|
|
| 27 |
A |
172 |
170 |
0.01 |
|
|
|
| 28 |
A |
116 |
115 |
0.00 |
|
|
|
| 29 |
A |
50 |
49 |
0.00 |
|
|
|
| 30 |
B |
3081 |
3047 |
22.75 |
|
|
|
| 31 |
B |
3062 |
3028 |
8.81 |
|
|
|
| 32 |
B |
3024 |
2991 |
15.21 |
|
|
|
| 33 |
B |
2977 |
2945 |
70.89 |
|
|
|
| 34 |
B |
2974 |
2942 |
15.90 |
|
|
|
| 35 |
B |
2886 |
2854 |
102.02 |
|
|
|
| 36 |
B |
2844 |
2813 |
31.14 |
|
|
|
| 37 |
B |
1412 |
1396 |
7.93 |
|
|
|
| 38 |
B |
1405 |
1389 |
12.71 |
|
|
|
| 39 |
B |
1388 |
1373 |
10.21 |
|
|
|
| 40 |
B |
1375 |
1360 |
5.25 |
|
|
|
| 41 |
B |
1334 |
1319 |
10.98 |
|
|
|
| 42 |
B |
1319 |
1304 |
44.88 |
|
|
|
| 43 |
B |
1285 |
1270 |
12.50 |
|
|
|
| 44 |
B |
1242 |
1228 |
24.11 |
|
|
|
| 45 |
B |
1208 |
1195 |
51.43 |
|
|
|
| 46 |
B |
1197 |
1183 |
117.55 |
|
|
|
| 47 |
B |
1129 |
1116 |
11.85 |
|
|
|
| 48 |
B |
1091 |
1079 |
3.66 |
|
|
|
| 49 |
B |
1001 |
990 |
36.28 |
|
|
|
| 50 |
B |
902 |
892 |
0.60 |
|
|
|
| 51 |
B |
885 |
875 |
1.99 |
|
|
|
| 52 |
B |
754 |
746 |
6.95 |
|
|
|
| 53 |
B |
490 |
484 |
1.05 |
|
|
|
| 54 |
B |
318 |
315 |
0.88 |
|
|
|
| 55 |
B |
226 |
224 |
3.68 |
|
|
|
| 56 |
B |
176 |
174 |
2.40 |
|
|
|
| 57 |
B |
75 |
75 |
0.88 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 41092.8 cm
-1
Scaled (by 0.989) Zero Point Vibrational Energy (zpe) 40640.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/cc-pVDZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.101 |
| C2 |
0.042 |
1.160 |
0.878 |
| C3 |
-0.042 |
-1.160 |
0.878 |
| C4 |
0.000 |
2.353 |
-0.038 |
| C5 |
0.000 |
-2.353 |
-0.038 |
| C6 |
-1.264 |
2.378 |
-0.865 |
| C7 |
1.264 |
-2.378 |
-0.865 |
| H8 |
-0.835 |
1.181 |
1.581 |
| H9 |
0.964 |
1.167 |
1.517 |
| H10 |
0.835 |
-1.181 |
1.581 |
| H11 |
-0.964 |
-1.167 |
1.517 |
| H12 |
0.894 |
2.313 |
-0.700 |
| H13 |
0.101 |
3.275 |
0.575 |
| H14 |
-0.894 |
-2.313 |
-0.700 |
| H15 |
-0.101 |
-3.275 |
0.575 |
| H16 |
-1.269 |
3.207 |
-1.600 |
| H17 |
-1.369 |
1.419 |
-1.412 |
| H18 |
-2.162 |
2.490 |
-0.219 |
| H19 |
1.269 |
-3.207 |
-1.600 |
| H20 |
1.369 |
-1.419 |
-1.412 |
| H21 |
2.162 |
-2.490 |
-0.219 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.3972 | 1.3972 | 2.3566 | 2.3566 | 2.8605 | 2.8605 | 2.0701 | 2.0727 | 2.0701 | 2.0727 | 2.6052 | 3.3103 | 2.6052 | 3.3103 | 3.8453 | 2.4858 | 3.3132 | 3.8453 | 2.4858 | 3.3132 |
C2 | 1.3972 | | 2.3221 | 1.5039 | 3.6305 | 2.4947 | 4.1288 | 1.1245 | 1.1216 | 2.5705 | 2.6145 | 2.1312 | 2.1368 | 3.9275 | 4.4476 | 3.4709 | 2.7027 | 2.7979 | 5.1689 | 3.6962 | 4.3618 | C3 | 1.3972 | 2.3221 | | 3.6305 | 1.5039 | 4.1288 | 2.4947 | 2.5705 | 2.6145 | 1.1245 | 1.1216 | 3.9275 | 4.4476 | 2.1312 | 2.1368 | 5.1689 | 3.6962 | 4.3618 | 3.4709 | 2.7027 | 2.7979 | C4 | 2.3566 | 1.5039 | 3.6305 | | 4.7052 | 1.5106 | 4.9655 | 2.1658 | 2.1798 | 3.9759 | 3.9665 | 1.1128 | 1.1116 | 4.7959 | 5.6614 | 2.1867 | 2.1533 | 2.1737 | 5.9124 | 4.2416 | 5.3066 | C5 | 2.3566 | 3.6305 | 1.5039 | 4.7052 | | 4.9655 | 1.5106 | 3.9759 | 3.9665 | 2.1658 | 2.1798 | 4.7959 | 5.6614 | 1.1128 | 1.1116 | 5.9124 | 4.2416 | 5.3066 | 2.1867 | 2.1533 | 2.1737 | C6 | 2.8605 | 2.4947 | 4.1288 | 1.5106 | 4.9655 | | 5.3854 | 2.7563 | 3.4785 | 4.8016 | 4.2810 | 2.1649 | 2.1772 | 4.7077 | 5.9475 | 1.1083 | 1.1091 | 1.1115 | 6.1758 | 4.6530 | 5.9874 | C7 | 2.8605 | 4.1288 | 2.4947 | 4.9655 | 1.5106 | 5.3854 | | 4.8016 | 4.2810 | 2.7563 | 3.4785 | 4.7077 | 5.9475 | 2.1649 | 2.1772 | 6.1758 | 4.6530 | 5.9874 | 1.1083 | 1.1091 | 1.1115 | H8 | 2.0701 | 1.1245 | 2.5705 | 2.1658 | 3.9759 | 2.7563 | 4.8016 | | 1.8000 | 2.8936 | 2.3527 | 3.0776 | 2.5043 | 4.1729 | 4.6268 | 3.7963 | 3.0503 | 2.5912 | 5.8141 | 4.5371 | 5.0700 | H9 | 2.0727 | 1.1216 | 2.6145 | 2.1798 | 3.9665 | 3.4785 | 4.2810 | 1.8000 | | 2.3527 | 3.0266 | 2.4963 | 2.4645 | 4.5244 | 4.6636 | 4.3431 | 3.7535 | 3.8122 | 5.3795 | 3.9287 | 4.2221 | H10 | 2.0701 | 2.5705 | 1.1245 | 3.9759 | 2.1658 | 4.8016 | 2.7563 | 2.8936 | 2.3527 | | 1.8000 | 4.1729 | 4.6268 | 3.0776 | 2.5043 | 5.8141 | 4.5371 | 5.0700 | 3.7963 | 3.0503 | 2.5912 | H11 | 2.0727 | 2.6145 | 1.1216 | 3.9665 | 2.1798 | 4.2810 | 3.4785 | 2.3527 | 3.0266 | 1.8000 | | 4.5244 | 4.6636 | 2.4963 | 2.4645 | 5.3795 | 3.9287 | 4.2221 | 4.3431 | 3.7535 | 3.8122 | H12 | 2.6052 | 2.1312 | 3.9275 | 1.1128 | 4.7959 | 2.1649 | 4.7077 | 3.0776 | 2.4963 | 4.1729 | 4.5244 | | 1.7827 | 4.9585 | 5.8164 | 2.5076 | 2.5354 | 3.0980 | 5.6047 | 3.8288 | 4.9907 | H13 | 3.3103 | 2.1368 | 4.4476 | 1.1116 | 5.6614 | 2.1772 | 5.9475 | 2.5043 | 2.4645 | 4.6268 | 4.6636 | 1.7827 | | 5.8164 | 6.5524 | 2.5717 | 3.0912 | 2.5231 | 6.9355 | 5.2526 | 6.1735 | H14 | 2.6052 | 3.9275 | 2.1312 | 4.7959 | 1.1128 | 4.7077 | 2.1649 | 4.1729 | 4.5244 | 3.0776 | 2.4963 | 4.9585 | 5.8164 | | 1.7827 | 5.6047 | 3.8288 | 4.9907 | 2.5076 | 2.5354 | 3.0980 | H15 | 3.3103 | 4.4476 | 2.1368 | 5.6614 | 1.1116 | 5.9475 | 2.1772 | 4.6268 | 4.6636 | 2.5043 | 2.4645 | 5.8164 | 6.5524 | 1.7827 | | 6.9355 | 5.2526 | 6.1735 | 2.5717 | 3.0912 | 2.5231 | H16 | 3.8453 | 3.4709 | 5.1689 | 2.1867 | 5.9124 | 1.1083 | 6.1758 | 3.7963 | 4.3431 | 5.8141 | 5.3795 | 2.5076 | 2.5717 | 5.6047 | 6.9355 | | 1.8001 | 1.7935 | 6.8972 | 5.3286 | 6.7922 | H17 | 2.4858 | 2.7027 | 3.6962 | 2.1533 | 4.2416 | 1.1091 | 4.6530 | 3.0503 | 3.7535 | 4.5371 | 3.9287 | 2.5354 | 3.0912 | 3.8288 | 5.2526 | 1.8001 | | 1.7886 | 5.3286 | 3.9443 | 5.4016 | H18 | 3.3132 | 2.7979 | 4.3618 | 2.1737 | 5.3066 | 1.1115 | 5.9874 | 2.5912 | 3.8122 | 5.0700 | 4.2221 | 3.0980 | 2.5231 | 4.9907 | 6.1735 | 1.7935 | 1.7886 | | 6.7922 | 5.4016 | 6.5954 | H19 | 3.8453 | 5.1689 | 3.4709 | 5.9124 | 2.1867 | 6.1758 | 1.1083 | 5.8141 | 5.3795 | 3.7963 | 4.3431 | 5.6047 | 6.9355 | 2.5076 | 2.5717 | 6.8972 | 5.3286 | 6.7922 | | 1.8001 | 1.7935 | H20 | 2.4858 | 3.6962 | 2.7027 | 4.2416 | 2.1533 | 4.6530 | 1.1091 | 4.5371 | 3.9287 | 3.0503 | 3.7535 | 3.8288 | 5.2526 | 2.5354 | 3.0912 | 5.3286 | 3.9443 | 5.4016 | 1.8001 | | 1.7886 | H21 | 3.3132 | 4.3618 | 2.7979 | 5.3066 | 2.1737 | 5.9874 | 1.1115 | 5.0700 | 4.2221 | 2.5912 | 3.8122 | 4.9907 | 6.1735 | 3.0980 | 2.5231 | 6.7922 | 5.4016 | 6.5954 | 1.7935 | 1.7886 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.593 |
|
O1 |
C2 |
H8 |
109.882 |
| O1 |
C2 |
H9 |
110.274 |
|
O1 |
C3 |
C5 |
108.593 |
| O1 |
C3 |
H10 |
109.882 |
|
O1 |
C3 |
H11 |
110.274 |
| C2 |
O1 |
C3 |
112.403 |
|
C2 |
C4 |
C6 |
111.699 |
| C2 |
C4 |
H12 |
108.139 |
|
C2 |
C4 |
H13 |
108.633 |
| C3 |
C5 |
C7 |
111.699 |
|
C3 |
C5 |
H14 |
108.139 |
| C3 |
C5 |
H15 |
108.633 |
|
C4 |
C2 |
H8 |
110.138 |
| C4 |
C2 |
H9 |
111.418 |
|
C4 |
C6 |
H16 |
112.315 |
| C4 |
C6 |
H17 |
109.614 |
|
C4 |
C6 |
H18 |
111.078 |
| C5 |
C3 |
H10 |
110.138 |
|
C5 |
C3 |
H11 |
111.418 |
| C5 |
C7 |
H19 |
112.315 |
|
C5 |
C7 |
H20 |
109.614 |
| C5 |
C7 |
H21 |
111.078 |
|
C6 |
C4 |
H12 |
110.298 |
| C6 |
C4 |
H13 |
111.350 |
|
C7 |
C5 |
H14 |
110.298 |
| C7 |
C5 |
H15 |
111.350 |
|
H8 |
C2 |
H9 |
106.526 |
| H10 |
C3 |
H11 |
106.526 |
|
H12 |
C4 |
H13 |
106.531 |
| H14 |
C5 |
H15 |
106.531 |
|
H16 |
C6 |
H17 |
108.549 |
| H16 |
C6 |
H18 |
107.803 |
|
H17 |
C6 |
H18 |
107.318 |
| H19 |
C7 |
H20 |
108.549 |
|
H19 |
C7 |
H21 |
107.803 |
| H20 |
C7 |
H21 |
107.318 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.223 |
|
|
|
| 2 |
C |
0.020 |
|
|
|
| 3 |
C |
0.020 |
|
|
|
| 4 |
C |
-0.134 |
|
|
|
| 5 |
C |
-0.134 |
|
|
|
| 6 |
C |
-0.158 |
|
|
|
| 7 |
C |
-0.158 |
|
|
|
| 8 |
H |
0.032 |
|
|
|
| 9 |
H |
0.040 |
|
|
|
| 10 |
H |
0.032 |
|
|
|
| 11 |
H |
0.040 |
|
|
|
| 12 |
H |
0.065 |
|
|
|
| 13 |
H |
0.054 |
|
|
|
| 14 |
H |
0.065 |
|
|
|
| 15 |
H |
0.054 |
|
|
|
| 16 |
H |
0.061 |
|
|
|
| 17 |
H |
0.073 |
|
|
|
| 18 |
H |
0.059 |
|
|
|
| 19 |
H |
0.061 |
|
|
|
| 20 |
H |
0.073 |
|
|
|
| 21 |
H |
0.059 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.799 |
0.799 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.230 |
0.355 |
0.000 |
| y |
0.355 |
-44.304 |
0.000 |
| z |
0.000 |
0.000 |
-46.348 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.904 |
0.355 |
0.000 |
| y |
0.355 |
2.485 |
0.000 |
| z |
0.000 |
0.000 |
-0.582 |
|
| Polar |
|---|
| 3z2-r2 | -1.163 |
|---|
| x2-y2 | -2.926 |
|---|
| xy | 0.355 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.662 |
-0.566 |
0.000 |
| y |
-0.566 |
12.868 |
0.000 |
| z |
0.000 |
0.000 |
10.764 |
<r2> (average value of r
2) Å
2
| <r2> |
332.918 |
| (<r2>)1/2 |
18.246 |