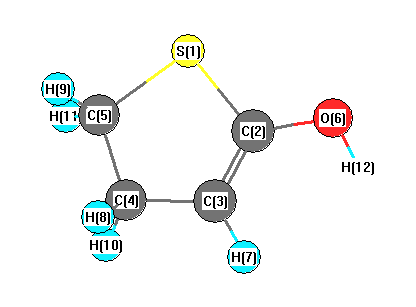
Vibrational Frequencies calculated at LSDA/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3700 |
3660 |
60.44 |
|
|
|
| 2 |
A |
3118 |
3084 |
6.12 |
|
|
|
| 3 |
A |
3061 |
3027 |
4.88 |
|
|
|
| 4 |
A |
3001 |
2968 |
21.46 |
|
|
|
| 5 |
A |
2979 |
2946 |
12.20 |
|
|
|
| 6 |
A |
2903 |
2871 |
47.08 |
|
|
|
| 7 |
A |
1667 |
1649 |
160.57 |
|
|
|
| 8 |
A |
1409 |
1394 |
1.70 |
|
|
|
| 9 |
A |
1390 |
1375 |
10.67 |
|
|
|
| 10 |
A |
1346 |
1331 |
17.00 |
|
|
|
| 11 |
A |
1283 |
1269 |
8.21 |
|
|
|
| 12 |
A |
1254 |
1241 |
46.36 |
|
|
|
| 13 |
A |
1229 |
1215 |
7.24 |
|
|
|
| 14 |
A |
1149 |
1137 |
19.01 |
|
|
|
| 15 |
A |
1109 |
1097 |
11.22 |
|
|
|
| 16 |
A |
1095 |
1083 |
150.31 |
|
|
|
| 17 |
A |
1017 |
1006 |
22.37 |
|
|
|
| 18 |
A |
963 |
953 |
5.75 |
|
|
|
| 19 |
A |
931 |
921 |
17.01 |
|
|
|
| 20 |
A |
845 |
836 |
2.36 |
|
|
|
| 21 |
A |
697 |
689 |
22.99 |
|
|
|
| 22 |
A |
692 |
684 |
26.60 |
|
|
|
| 23 |
A |
682 |
675 |
13.46 |
|
|
|
| 24 |
A |
616 |
609 |
5.30 |
|
|
|
| 25 |
A |
518 |
512 |
9.66 |
|
|
|
| 26 |
A |
461 |
456 |
2.75 |
|
|
|
| 27 |
A |
357 |
353 |
28.28 |
|
|
|
| 28 |
A |
351 |
347 |
62.77 |
|
|
|
| 29 |
A |
257 |
254 |
8.58 |
|
|
|
| 30 |
A |
179 |
177 |
2.81 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20129.3 cm
-1
Scaled (by 0.9891) Zero Point Vibrational Energy (zpe) 19909.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.006 |
|
|
|
| 2 |
C |
0.073 |
|
|
|
| 3 |
C |
-0.253 |
|
|
|
| 4 |
C |
-0.202 |
|
|
|
| 5 |
C |
-0.242 |
|
|
|
| 6 |
O |
-0.208 |
|
|
|
| 7 |
H |
0.117 |
|
|
|
| 8 |
H |
0.115 |
|
|
|
| 9 |
H |
0.137 |
|
|
|
| 10 |
H |
0.113 |
|
|
|
| 11 |
H |
0.127 |
|
|
|
| 12 |
H |
0.217 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.473 |
2.210 |
0.080 |
2.262 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.254 |
-4.751 |
0.756 |
| y |
-4.751 |
-42.339 |
-0.150 |
| z |
0.756 |
-0.150 |
-45.225 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.528 |
-4.751 |
0.756 |
| y |
-4.751 |
-1.099 |
-0.150 |
| z |
0.756 |
-0.150 |
-5.429 |
|
| Polar |
|---|
| 3z2-r2 | -10.858 |
|---|
| x2-y2 | 5.085 |
|---|
| xy | -4.751 |
|---|
| xz | 0.756 |
|---|
| yz | -0.150 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.510 |
0.277 |
0.018 |
| y |
0.277 |
11.568 |
0.153 |
| z |
0.018 |
0.153 |
6.927 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |