Jump to
S1C2
Energy calculated at LSDA/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -340.791466 |
| Energy at 298.15K | |
| HF Energy | -340.791466 |
| Nuclear repulsion energy | 247.015013 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2971 |
2939 |
50.05 |
|
|
|
| 2 |
A1 |
1929 |
1908 |
577.37 |
|
|
|
| 3 |
A1 |
1475 |
1459 |
0.13 |
|
|
|
| 4 |
A1 |
1332 |
1317 |
0.16 |
|
|
|
| 5 |
A1 |
1206 |
1193 |
218.03 |
|
|
|
| 6 |
A1 |
953 |
942 |
5.35 |
|
|
|
| 7 |
A1 |
846 |
837 |
31.78 |
|
|
|
| 8 |
A1 |
722 |
714 |
0.43 |
|
|
|
| 9 |
A2 |
2996 |
2963 |
0.00 |
|
|
|
| 10 |
A2 |
1204 |
1191 |
0.00 |
|
|
|
| 11 |
A2 |
1122 |
1110 |
0.00 |
|
|
|
| 12 |
A2 |
174i |
173i |
0.00 |
|
|
|
| 13 |
B1 |
3018 |
2985 |
25.25 |
|
|
|
| 14 |
B1 |
1222 |
1209 |
0.96 |
|
|
|
| 15 |
B1 |
832 |
823 |
0.15 |
|
|
|
| 16 |
B1 |
747 |
739 |
15.98 |
|
|
|
| 17 |
B1 |
156 |
154 |
1.23 |
|
|
|
| 18 |
B2 |
2967 |
2935 |
18.87 |
|
|
|
| 19 |
B2 |
1463 |
1447 |
8.43 |
|
|
|
| 20 |
B2 |
1351 |
1337 |
9.95 |
|
|
|
| 21 |
B2 |
1194 |
1181 |
7.34 |
|
|
|
| 22 |
B2 |
1036 |
1025 |
287.74 |
|
|
|
| 23 |
B2 |
744 |
736 |
1.17 |
|
|
|
| 24 |
B2 |
499 |
494 |
0.12 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15905.8 cm
-1
Scaled (by 0.9891) Zero Point Vibrational Energy (zpe) 15732.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.860 |
| O2 |
0.000 |
0.000 |
2.042 |
| O3 |
0.000 |
1.120 |
0.056 |
| O4 |
0.000 |
-1.120 |
0.056 |
| C5 |
0.000 |
0.767 |
-1.277 |
| C6 |
0.000 |
-0.767 |
-1.277 |
| H7 |
-0.892 |
1.184 |
-1.770 |
| H8 |
0.892 |
1.184 |
-1.770 |
| H9 |
0.892 |
-1.184 |
-1.770 |
| H10 |
-0.892 |
-1.184 |
-1.770 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1818 | 1.3786 | 1.3786 | 2.2704 | 2.2704 | 3.0192 | 3.0192 | 3.0192 | 3.0192 |
O2 | 1.1818 | | 2.2799 | 2.2799 | 3.4062 | 3.4062 | 4.0902 | 4.0902 | 4.0902 | 4.0902 | O3 | 1.3786 | 2.2799 | | 2.2395 | 1.3786 | 2.3101 | 2.0334 | 2.0334 | 3.0718 | 3.0718 | O4 | 1.3786 | 2.2799 | 2.2395 | | 2.3101 | 1.3786 | 3.0718 | 3.0718 | 2.0334 | 2.0334 | C5 | 2.2704 | 3.4062 | 1.3786 | 2.3101 | | 1.5342 | 1.1012 | 1.1012 | 2.2010 | 2.2010 | C6 | 2.2704 | 3.4062 | 2.3101 | 1.3786 | 1.5342 | | 2.2010 | 2.2010 | 1.1012 | 1.1012 | H7 | 3.0192 | 4.0902 | 2.0334 | 3.0718 | 1.1012 | 2.2010 | | 1.7840 | 2.9642 | 2.3672 | H8 | 3.0192 | 4.0902 | 2.0334 | 3.0718 | 1.1012 | 2.2010 | 1.7840 | | 2.3672 | 2.9642 | H9 | 3.0192 | 4.0902 | 3.0718 | 2.0334 | 2.2010 | 1.1012 | 2.9642 | 2.3672 | | 1.7840 | H10 | 3.0192 | 4.0902 | 3.0718 | 2.0334 | 2.2010 | 1.1012 | 2.3672 | 2.9642 | 1.7840 | |
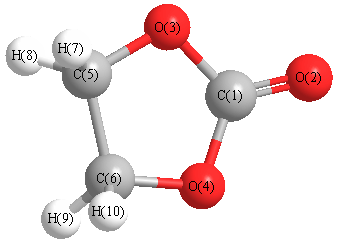 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.864 |
|
C1 |
O4 |
C6 |
110.864 |
| O2 |
C1 |
O3 |
125.685 |
|
O2 |
C1 |
O4 |
125.685 |
| O3 |
C1 |
O4 |
108.630 |
|
O3 |
C5 |
C6 |
104.821 |
| O3 |
C5 |
H7 |
109.662 |
|
O3 |
C5 |
H8 |
109.662 |
| O4 |
C6 |
C5 |
104.821 |
|
O4 |
C6 |
H9 |
109.662 |
| O4 |
C6 |
H10 |
109.662 |
|
C5 |
C6 |
H9 |
112.223 |
| C5 |
C6 |
H10 |
112.223 |
|
C6 |
C5 |
H7 |
112.223 |
| C6 |
C5 |
H8 |
112.223 |
|
H7 |
C5 |
H8 |
108.197 |
| H9 |
C6 |
H10 |
108.197 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.237 |
|
|
|
| 2 |
O |
-0.208 |
|
|
|
| 3 |
O |
-0.161 |
|
|
|
| 4 |
O |
-0.161 |
|
|
|
| 5 |
C |
-0.049 |
|
|
|
| 6 |
C |
-0.049 |
|
|
|
| 7 |
H |
0.098 |
|
|
|
| 8 |
H |
0.098 |
|
|
|
| 9 |
H |
0.098 |
|
|
|
| 10 |
H |
0.098 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.744 |
4.744 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.266 |
0.000 |
0.000 |
| y |
0.000 |
-36.016 |
0.000 |
| z |
0.000 |
0.000 |
-36.089 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.786 |
0.000 |
0.000 |
| y |
0.000 |
-1.839 |
0.000 |
| z |
0.000 |
0.000 |
-1.947 |
|
| Polar |
|---|
| 3z2-r2 | -3.894 |
|---|
| x2-y2 | 3.750 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.878 |
0.000 |
0.000 |
| y |
0.000 |
6.032 |
0.000 |
| z |
0.000 |
0.000 |
8.206 |
<r2> (average value of r
2) Å
2
| <r2> |
127.581 |
| (<r2>)1/2 |
11.295 |
Jump to
S1C1
Energy calculated at LSDA/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -340.792329 |
| Energy at 298.15K | -340.798704 |
| HF Energy | -340.792329 |
| Nuclear repulsion energy | 247.648403 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3031 |
2998 |
13.88 |
|
|
|
| 2 |
A |
2942 |
2910 |
26.02 |
|
|
|
| 3 |
A |
1932 |
1911 |
560.38 |
|
|
|
| 4 |
A |
1457 |
1441 |
0.35 |
|
|
|
| 5 |
A |
1336 |
1322 |
2.77 |
|
|
|
| 6 |
A |
1215 |
1202 |
66.84 |
|
|
|
| 7 |
A |
1196 |
1183 |
126.87 |
|
|
|
| 8 |
A |
1109 |
1097 |
18.78 |
|
|
|
| 9 |
A |
955 |
945 |
1.24 |
|
|
|
| 10 |
A |
839 |
830 |
34.57 |
|
|
|
| 11 |
A |
719 |
711 |
0.60 |
|
|
|
| 12 |
A |
148 |
146 |
0.33 |
|
|
|
| 13 |
B |
3040 |
3007 |
11.15 |
|
|
|
| 14 |
B |
2951 |
2919 |
38.83 |
|
|
|
| 15 |
B |
1450 |
1434 |
10.68 |
|
|
|
| 16 |
B |
1342 |
1327 |
8.11 |
|
|
|
| 17 |
B |
1229 |
1216 |
4.35 |
|
|
|
| 18 |
B |
1160 |
1147 |
4.82 |
|
|
|
| 19 |
B |
1027 |
1015 |
267.01 |
|
|
|
| 20 |
B |
879 |
869 |
5.24 |
|
|
|
| 21 |
B |
755 |
747 |
16.63 |
|
|
|
| 22 |
B |
666 |
659 |
0.28 |
|
|
|
| 23 |
B |
499 |
494 |
0.26 |
|
|
|
| 24 |
B |
160 |
159 |
1.09 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16017.7 cm
-1
Scaled (by 0.9891) Zero Point Vibrational Energy (zpe) 15843.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.855 |
| O2 |
0.000 |
0.000 |
2.037 |
| O3 |
0.000 |
1.120 |
0.045 |
| O4 |
0.000 |
-1.120 |
0.045 |
| C5 |
0.201 |
0.731 |
-1.266 |
| C6 |
-0.201 |
-0.731 |
-1.266 |
| H7 |
-0.415 |
1.346 |
-1.937 |
| H8 |
1.264 |
0.865 |
-1.536 |
| H9 |
0.415 |
-1.346 |
-1.937 |
| H10 |
-1.264 |
-0.865 |
-1.536 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1815 | 1.3824 | 1.3824 | 2.2529 | 2.2529 | 3.1272 | 2.8399 | 3.1272 | 2.8399 |
O2 | 1.1815 | | 2.2849 | 2.2849 | 3.3888 | 3.3888 | 4.2157 | 3.8874 | 4.2157 | 3.8874 | O3 | 1.3824 | 2.2849 | | 2.2402 | 1.3824 | 2.2777 | 2.0374 | 2.0402 | 3.1910 | 2.8355 | O4 | 1.3824 | 2.2849 | 2.2402 | | 2.2777 | 1.3824 | 3.1910 | 2.8355 | 2.0374 | 2.0402 | C5 | 2.2529 | 3.3888 | 1.3824 | 2.2777 | | 1.5169 | 1.0984 | 1.1047 | 2.1935 | 2.1834 | C6 | 2.2529 | 3.3888 | 2.2777 | 1.3824 | 1.5169 | | 2.1935 | 2.1834 | 1.0984 | 1.1047 | H7 | 3.1272 | 4.2157 | 2.0374 | 3.1910 | 1.0984 | 2.1935 | | 1.7910 | 2.8170 | 2.4025 | H8 | 2.8399 | 3.8874 | 2.0402 | 2.8355 | 1.1047 | 2.1834 | 1.7910 | | 2.4025 | 3.0630 | H9 | 3.1272 | 4.2157 | 3.1910 | 2.0374 | 2.1935 | 1.0984 | 2.8170 | 2.4025 | | 1.7910 | H10 | 2.8399 | 3.8874 | 2.8355 | 2.0402 | 2.1834 | 1.1047 | 2.4025 | 3.0630 | 1.7910 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
109.149 |
|
C1 |
O4 |
C6 |
109.149 |
| O2 |
C1 |
O3 |
125.875 |
|
O2 |
C1 |
O4 |
125.875 |
| O3 |
C1 |
O4 |
108.251 |
|
O3 |
C5 |
C6 |
103.457 |
| O3 |
C5 |
H7 |
109.903 |
|
O3 |
C5 |
H8 |
109.731 |
| O4 |
C6 |
C5 |
103.457 |
|
O4 |
C6 |
H9 |
109.903 |
| O4 |
C6 |
H10 |
109.731 |
|
C5 |
C6 |
H9 |
113.038 |
| C5 |
C6 |
H10 |
111.821 |
|
C6 |
C5 |
H7 |
113.038 |
| C6 |
C5 |
H8 |
111.821 |
|
H7 |
C5 |
H8 |
108.772 |
| H9 |
C6 |
H10 |
108.772 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.240 |
|
|
|
| 2 |
O |
-0.208 |
|
|
|
| 3 |
O |
-0.165 |
|
|
|
| 4 |
O |
-0.165 |
|
|
|
| 5 |
C |
-0.054 |
|
|
|
| 6 |
C |
-0.054 |
|
|
|
| 7 |
H |
0.106 |
|
|
|
| 8 |
H |
0.096 |
|
|
|
| 9 |
H |
0.106 |
|
|
|
| 10 |
H |
0.096 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.702 |
4.702 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.388 |
0.189 |
0.000 |
| y |
0.189 |
-36.035 |
0.000 |
| z |
0.000 |
0.000 |
-35.849 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.554 |
0.189 |
0.000 |
| y |
0.189 |
-1.916 |
0.000 |
| z |
0.000 |
0.000 |
-1.638 |
|
| Polar |
|---|
| 3z2-r2 | -3.276 |
|---|
| x2-y2 | 3.647 |
|---|
| xy | 0.189 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.014 |
0.202 |
0.000 |
| y |
0.202 |
5.954 |
0.000 |
| z |
0.000 |
0.000 |
8.157 |
<r2> (average value of r
2) Å
2
| <r2> |
126.412 |
| (<r2>)1/2 |
11.243 |