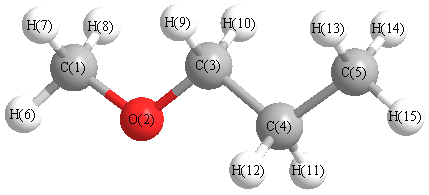
Vibrational Frequencies calculated at LSDA/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3073 |
3018 |
15.57 |
|
|
|
| 2 |
A' |
3065 |
3010 |
17.80 |
|
|
|
| 3 |
A' |
3002 |
2948 |
15.93 |
|
|
|
| 4 |
A' |
2980 |
2926 |
13.34 |
|
|
|
| 5 |
A' |
2890 |
2838 |
60.80 |
|
|
|
| 6 |
A' |
2859 |
2807 |
37.60 |
|
|
|
| 7 |
A' |
1531 |
1504 |
3.75 |
|
|
|
| 8 |
A' |
1510 |
1483 |
22.18 |
|
|
|
| 9 |
A' |
1503 |
1475 |
2.91 |
|
|
|
| 10 |
A' |
1489 |
1463 |
6.77 |
|
|
|
| 11 |
A' |
1452 |
1426 |
1.49 |
|
|
|
| 12 |
A' |
1403 |
1377 |
23.87 |
|
|
|
| 13 |
A' |
1374 |
1349 |
8.04 |
|
|
|
| 14 |
A' |
1307 |
1284 |
0.16 |
|
|
|
| 15 |
A' |
1181 |
1160 |
30.24 |
|
|
|
| 16 |
A' |
1147 |
1126 |
89.86 |
|
|
|
| 17 |
A' |
1114 |
1094 |
20.42 |
|
|
|
| 18 |
A' |
1065 |
1046 |
0.52 |
|
|
|
| 19 |
A' |
966 |
948 |
9.03 |
|
|
|
| 20 |
A' |
904 |
888 |
19.62 |
|
|
|
| 21 |
A' |
432 |
424 |
1.71 |
|
|
|
| 22 |
A' |
403 |
396 |
3.28 |
|
|
|
| 23 |
A' |
180 |
177 |
2.18 |
|
|
|
| 24 |
A" |
3064 |
3009 |
31.55 |
|
|
|
| 25 |
A" |
3037 |
2982 |
0.03 |
|
|
|
| 26 |
A" |
2927 |
2874 |
56.58 |
|
|
|
| 27 |
A" |
2883 |
2831 |
68.61 |
|
|
|
| 28 |
A" |
1502 |
1475 |
15.52 |
|
|
|
| 29 |
A" |
1476 |
1450 |
13.37 |
|
|
|
| 30 |
A" |
1300 |
1276 |
0.03 |
|
|
|
| 31 |
A" |
1241 |
1219 |
0.97 |
|
|
|
| 32 |
A" |
1173 |
1152 |
1.54 |
|
|
|
| 33 |
A" |
1131 |
1110 |
0.00 |
|
|
|
| 34 |
A" |
910 |
894 |
2.78 |
|
|
|
| 35 |
A" |
780 |
766 |
6.54 |
|
|
|
| 36 |
A" |
258 |
253 |
2.31 |
|
|
|
| 37 |
A" |
244 |
240 |
0.93 |
|
|
|
| 38 |
A" |
124 |
122 |
4.33 |
|
|
|
| 39 |
A" |
106 |
104 |
0.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29492.2 cm
-1
Scaled (by 0.982) Zero Point Vibrational Energy (zpe) 28961.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.408 |
|
|
|
| 2 |
O |
-0.429 |
|
|
|
| 3 |
C |
-0.159 |
|
|
|
| 4 |
C |
-0.457 |
|
|
|
| 5 |
C |
-0.637 |
|
|
|
| 6 |
H |
0.230 |
|
|
|
| 7 |
H |
0.193 |
|
|
|
| 8 |
H |
0.193 |
|
|
|
| 9 |
H |
0.186 |
|
|
|
| 10 |
H |
0.186 |
|
|
|
| 11 |
H |
0.228 |
|
|
|
| 12 |
H |
0.228 |
|
|
|
| 13 |
H |
0.212 |
|
|
|
| 14 |
H |
0.212 |
|
|
|
| 15 |
H |
0.221 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.150 |
1.190 |
0.000 |
1.199 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.123 |
-2.180 |
0.000 |
| y |
-2.180 |
-33.424 |
0.000 |
| z |
0.000 |
0.000 |
-33.124 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.151 |
-2.180 |
0.000 |
| y |
-2.180 |
-1.801 |
0.000 |
| z |
0.000 |
0.000 |
-1.350 |
|
| Polar |
|---|
| 3z2-r2 | -2.700 |
|---|
| x2-y2 | 3.301 |
|---|
| xy | -2.180 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.397 |
-0.345 |
0.000 |
| y |
-0.345 |
6.279 |
0.000 |
| z |
0.000 |
0.000 |
6.142 |
<r2> (average value of r
2) Å
2
| <r2> |
179.988 |
| (<r2>)1/2 |
13.416 |