Vibrational Frequencies calculated at LSDA/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3420 |
3358 |
17.01 |
|
|
|
| 2 |
A |
3156 |
3099 |
4.77 |
|
|
|
| 3 |
A |
3087 |
3031 |
3.54 |
|
|
|
| 4 |
A |
3035 |
2980 |
15.12 |
|
|
|
| 5 |
A |
3008 |
2953 |
9.10 |
|
|
|
| 6 |
A |
2940 |
2887 |
33.04 |
|
|
|
| 7 |
A |
1668 |
1638 |
111.86 |
|
|
|
| 8 |
A |
1470 |
1444 |
1.09 |
|
|
|
| 9 |
A |
1454 |
1428 |
12.46 |
|
|
|
| 10 |
A |
1378 |
1353 |
19.29 |
|
|
|
| 11 |
A |
1309 |
1286 |
15.54 |
|
|
|
| 12 |
A |
1282 |
1259 |
12.72 |
|
|
|
| 13 |
A |
1261 |
1238 |
4.51 |
|
|
|
| 14 |
A |
1178 |
1156 |
3.47 |
|
|
|
| 15 |
A |
1150 |
1130 |
0.22 |
|
|
|
| 16 |
A |
1091 |
1071 |
171.45 |
|
|
|
| 17 |
A |
1034 |
1015 |
60.50 |
|
|
|
| 18 |
A |
980 |
963 |
16.62 |
|
|
|
| 19 |
A |
928 |
911 |
18.92 |
|
|
|
| 20 |
A |
864 |
848 |
5.47 |
|
|
|
| 21 |
A |
726 |
713 |
66.03 |
|
|
|
| 22 |
A |
690 |
678 |
6.95 |
|
|
|
| 23 |
A |
672 |
660 |
14.82 |
|
|
|
| 24 |
A |
624 |
613 |
8.14 |
|
|
|
| 25 |
A |
534 |
524 |
3.62 |
|
|
|
| 26 |
A |
470 |
462 |
3.05 |
|
|
|
| 27 |
A |
380 |
373 |
129.81 |
|
|
|
| 28 |
A |
342 |
336 |
10.28 |
|
|
|
| 29 |
A |
257 |
253 |
6.14 |
|
|
|
| 30 |
A |
168 |
165 |
2.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20276.6 cm
-1
Scaled (by 0.982) Zero Point Vibrational Energy (zpe) 19911.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
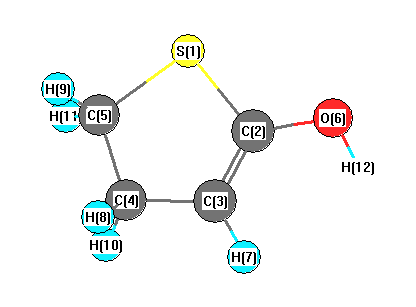
Charges from optimized geometry at LSDA/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.265 |
|
|
|
| 2 |
C |
0.045 |
|
|
|
| 3 |
C |
-0.224 |
|
|
|
| 4 |
C |
-0.481 |
|
|
|
| 5 |
C |
-0.604 |
|
|
|
| 6 |
O |
-0.532 |
|
|
|
| 7 |
H |
0.193 |
|
|
|
| 8 |
H |
0.229 |
|
|
|
| 9 |
H |
0.256 |
|
|
|
| 10 |
H |
0.233 |
|
|
|
| 11 |
H |
0.246 |
|
|
|
| 12 |
H |
0.374 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.411 |
2.572 |
0.116 |
2.607 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.192 |
-5.555 |
0.755 |
| y |
-5.555 |
-41.590 |
-0.026 |
| z |
0.755 |
-0.026 |
-45.029 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.117 |
-5.555 |
0.755 |
| y |
-5.555 |
-0.979 |
-0.026 |
| z |
0.755 |
-0.026 |
-6.138 |
|
| Polar |
|---|
| 3z2-r2 | -12.276 |
|---|
| x2-y2 | 5.397 |
|---|
| xy | -5.555 |
|---|
| xz | 0.755 |
|---|
| yz | -0.026 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.860 |
0.247 |
-0.012 |
| y |
0.247 |
9.598 |
0.176 |
| z |
-0.012 |
0.176 |
4.967 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |