Vibrational Frequencies calculated at SVWN/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3587 |
3519 |
65.06 |
|
|
|
| 2 |
A' |
3214 |
3152 |
0.79 |
|
|
|
| 3 |
A' |
3195 |
3135 |
0.55 |
|
|
|
| 4 |
A' |
3144 |
3084 |
11.90 |
|
|
|
| 5 |
A' |
3133 |
3073 |
19.58 |
|
|
|
| 6 |
A' |
3122 |
3063 |
1.65 |
|
|
|
| 7 |
A' |
3117 |
3058 |
1.99 |
|
|
|
| 8 |
A' |
1678 |
1646 |
2.05 |
|
|
|
| 9 |
A' |
1628 |
1597 |
2.48 |
|
|
|
| 10 |
A' |
1557 |
1527 |
5.36 |
|
|
|
| 11 |
A' |
1518 |
1490 |
7.34 |
|
|
|
| 12 |
A' |
1500 |
1472 |
12.23 |
|
|
|
| 13 |
A' |
1456 |
1428 |
5.74 |
|
|
|
| 14 |
A' |
1415 |
1388 |
43.74 |
|
|
|
| 15 |
A' |
1356 |
1330 |
8.72 |
|
|
|
| 16 |
A' |
1307 |
1282 |
3.13 |
|
|
|
| 17 |
A' |
1239 |
1216 |
9.46 |
|
|
|
| 18 |
A' |
1212 |
1189 |
3.43 |
|
|
|
| 19 |
A' |
1143 |
1121 |
0.65 |
|
|
|
| 20 |
A' |
1124 |
1102 |
1.27 |
|
|
|
| 21 |
A' |
1099 |
1078 |
21.99 |
|
|
|
| 22 |
A' |
1062 |
1042 |
5.14 |
|
|
|
| 23 |
A' |
1031 |
1012 |
5.51 |
|
|
|
| 24 |
A' |
893 |
876 |
6.13 |
|
|
|
| 25 |
A' |
865 |
848 |
0.42 |
|
|
|
| 26 |
A' |
771 |
757 |
2.06 |
|
|
|
| 27 |
A' |
603 |
591 |
1.17 |
|
|
|
| 28 |
A' |
541 |
531 |
0.10 |
|
|
|
| 29 |
A' |
392 |
384 |
3.54 |
|
|
|
| 30 |
A" |
928 |
910 |
0.08 |
|
|
|
| 31 |
A" |
882 |
865 |
2.50 |
|
|
|
| 32 |
A" |
836 |
820 |
4.12 |
|
|
|
| 33 |
A" |
814 |
798 |
3.25 |
|
|
|
| 34 |
A" |
766 |
752 |
1.95 |
|
|
|
| 35 |
A" |
727 |
713 |
81.06 |
|
|
|
| 36 |
A" |
702 |
688 |
32.23 |
|
|
|
| 37 |
A" |
612 |
600 |
6.92 |
|
|
|
| 38 |
A" |
576 |
565 |
0.67 |
|
|
|
| 39 |
A" |
437 |
429 |
43.56 |
|
|
|
| 40 |
A" |
418 |
410 |
41.10 |
|
|
|
| 41 |
A" |
244 |
239 |
0.16 |
|
|
|
| 42 |
A" |
215 |
211 |
9.10 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28027.4 cm
-1
Scaled (by 0.981) Zero Point Vibrational Energy (zpe) 27494.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
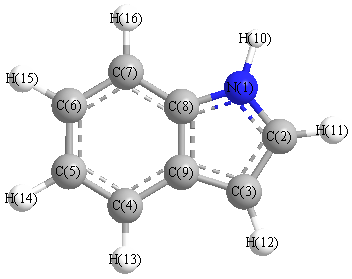
Charges from optimized geometry at SVWN/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.657 |
|
|
|
| 2 |
C |
-0.002 |
|
|
|
| 3 |
C |
-0.244 |
|
|
|
| 4 |
C |
-0.230 |
|
|
|
| 5 |
C |
-0.163 |
|
|
|
| 6 |
C |
-0.188 |
|
|
|
| 7 |
C |
-0.165 |
|
|
|
| 8 |
C |
0.275 |
|
|
|
| 9 |
C |
0.130 |
|
|
|
| 10 |
H |
0.343 |
|
|
|
| 11 |
H |
0.173 |
|
|
|
| 12 |
H |
0.153 |
|
|
|
| 13 |
H |
0.147 |
|
|
|
| 14 |
H |
0.142 |
|
|
|
| 15 |
H |
0.144 |
|
|
|
| 16 |
H |
0.142 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.925 |
2.099 |
0.000 |
2.294 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-45.780 |
-2.239 |
0.000 |
| y |
-2.239 |
-40.937 |
0.000 |
| z |
0.000 |
0.000 |
-56.460 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.919 |
-2.239 |
0.000 |
| y |
-2.239 |
10.183 |
0.000 |
| z |
0.000 |
0.000 |
-13.102 |
|
| Polar |
|---|
| 3z2-r2 | -26.204 |
|---|
| x2-y2 | -4.843 |
|---|
| xy | -2.239 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
18.177 |
-1.473 |
0.000 |
| y |
-1.473 |
14.533 |
0.000 |
| z |
0.000 |
0.000 |
4.425 |
<r2> (average value of r
2) Å
2
| <r2> |
279.959 |
| (<r2>)1/2 |
16.732 |