Vibrational Frequencies calculated at PBE1PBE/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3151 |
3030 |
35.36 |
|
|
|
| 2 |
A' |
3150 |
3029 |
11.86 |
|
|
|
| 3 |
A' |
3062 |
2945 |
16.77 |
|
|
|
| 4 |
A' |
2992 |
2877 |
90.86 |
|
|
|
| 5 |
A' |
2978 |
2863 |
38.27 |
|
|
|
| 6 |
A' |
1500 |
1442 |
3.67 |
|
|
|
| 7 |
A' |
1475 |
1418 |
3.03 |
|
|
|
| 8 |
A' |
1471 |
1415 |
5.39 |
|
|
|
| 9 |
A' |
1448 |
1393 |
2.43 |
|
|
|
| 10 |
A' |
1410 |
1356 |
29.99 |
|
|
|
| 11 |
A' |
1366 |
1313 |
0.65 |
|
|
|
| 12 |
A' |
1233 |
1186 |
67.25 |
|
|
|
| 13 |
A' |
1188 |
1143 |
120.26 |
|
|
|
| 14 |
A' |
1111 |
1068 |
3.25 |
|
|
|
| 15 |
A' |
1060 |
1019 |
21.08 |
|
|
|
| 16 |
A' |
876 |
842 |
8.85 |
|
|
|
| 17 |
A' |
469 |
451 |
0.59 |
|
|
|
| 18 |
A' |
289 |
278 |
2.67 |
|
|
|
| 19 |
A" |
3153 |
3032 |
24.80 |
|
|
|
| 20 |
A" |
3048 |
2931 |
52.38 |
|
|
|
| 21 |
A" |
3013 |
2898 |
64.69 |
|
|
|
| 22 |
A" |
1458 |
1402 |
10.80 |
|
|
|
| 23 |
A" |
1453 |
1397 |
3.97 |
|
|
|
| 24 |
A" |
1286 |
1237 |
1.82 |
|
|
|
| 25 |
A" |
1183 |
1138 |
8.50 |
|
|
|
| 26 |
A" |
1152 |
1108 |
0.07 |
|
|
|
| 27 |
A" |
814 |
782 |
0.54 |
|
|
|
| 28 |
A" |
256 |
246 |
1.70 |
|
|
|
| 29 |
A" |
205 |
197 |
1.27 |
|
|
|
| 30 |
A" |
116 |
112 |
2.47 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23681.7 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 22772.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
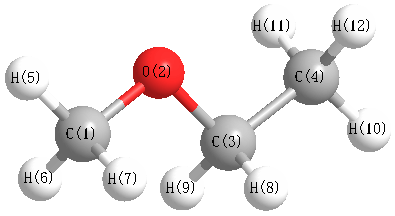
Charges from optimized geometry at PBE1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.750 |
|
|
|
| 2 |
O |
-0.604 |
|
|
|
| 3 |
C |
0.813 |
|
|
|
| 4 |
C |
0.345 |
|
|
|
| 5 |
H |
-0.313 |
|
|
|
| 6 |
H |
-0.094 |
|
|
|
| 7 |
H |
-0.094 |
|
|
|
| 8 |
H |
-0.248 |
|
|
|
| 9 |
H |
-0.248 |
|
|
|
| 10 |
H |
-0.087 |
|
|
|
| 11 |
H |
-0.110 |
|
|
|
| 12 |
H |
-0.110 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.891 |
-0.729 |
0.000 |
1.151 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.031 |
1.747 |
0.000 |
| y |
1.747 |
-25.922 |
0.000 |
| z |
0.000 |
0.000 |
-26.727 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.294 |
1.747 |
0.000 |
| y |
1.747 |
0.457 |
0.000 |
| z |
0.000 |
0.000 |
-0.750 |
|
| Polar |
|---|
| 3z2-r2 | -1.501 |
|---|
| x2-y2 | -0.109 |
|---|
| xy | 1.747 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.358 |
0.834 |
0.000 |
| y |
0.834 |
7.225 |
0.000 |
| z |
0.000 |
0.000 |
6.202 |
<r2> (average value of r
2) Å
2
| <r2> |
103.219 |
| (<r2>)1/2 |
10.160 |