Jump to
S1C2
Vibrational Frequencies calculated at PBE1PBE/aug-cc-pVDZ
Geometric Data calculated at PBE1PBE/aug-cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at PBE1PBE/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -323.324104 |
| Energy at 298.15K | -323.333146 |
| HF Energy | -323.324104 |
| Nuclear repulsion energy | 242.184623 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBE1PBE/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3148 |
3027 |
24.02 |
|
|
|
| 2 |
A |
3140 |
3019 |
35.17 |
|
|
|
| 3 |
A |
3133 |
3013 |
16.08 |
|
|
|
| 4 |
A |
3106 |
2986 |
11.66 |
|
|
|
| 5 |
A |
3069 |
2951 |
28.43 |
|
|
|
| 6 |
A |
3060 |
2943 |
17.38 |
|
|
|
| 7 |
A |
3056 |
2939 |
17.67 |
|
|
|
| 8 |
A |
1788 |
1719 |
255.48 |
|
|
|
| 9 |
A |
1477 |
1420 |
7.94 |
|
|
|
| 10 |
A |
1473 |
1417 |
11.05 |
|
|
|
| 11 |
A |
1462 |
1405 |
3.13 |
|
|
|
| 12 |
A |
1453 |
1397 |
2.54 |
|
|
|
| 13 |
A |
1399 |
1345 |
6.57 |
|
|
|
| 14 |
A |
1389 |
1336 |
7.02 |
|
|
|
| 15 |
A |
1359 |
1307 |
0.72 |
|
|
|
| 16 |
A |
1296 |
1246 |
7.56 |
|
|
|
| 17 |
A |
1280 |
1231 |
10.90 |
|
|
|
| 18 |
A |
1181 |
1135 |
12.07 |
|
|
|
| 19 |
A |
1117 |
1074 |
3.80 |
|
|
|
| 20 |
A |
1107 |
1065 |
29.04 |
|
|
|
| 21 |
A |
1014 |
975 |
5.52 |
|
|
|
| 22 |
A |
930 |
895 |
166.01 |
|
|
|
| 23 |
A |
892 |
857 |
11.78 |
|
|
|
| 24 |
A |
863 |
830 |
316.54 |
|
|
|
| 25 |
A |
779 |
749 |
10.35 |
|
|
|
| 26 |
A |
637 |
612 |
17.62 |
|
|
|
| 27 |
A |
458 |
440 |
4.70 |
|
|
|
| 28 |
A |
387 |
372 |
3.67 |
|
|
|
| 29 |
A |
286 |
275 |
0.66 |
|
|
|
| 30 |
A |
237 |
228 |
0.52 |
|
|
|
| 31 |
A |
184 |
177 |
0.43 |
|
|
|
| 32 |
A |
107 |
102 |
0.82 |
|
|
|
| 33 |
A |
51 |
49 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23157.6 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 22268.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBE1PBE/aug-cc-pVDZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.201 |
-0.911 |
0.181 |
| C2 |
-1.752 |
0.459 |
-0.315 |
| C3 |
-0.402 |
0.886 |
0.224 |
| O4 |
0.580 |
-0.034 |
-0.287 |
| N5 |
1.792 |
0.202 |
0.312 |
| O6 |
2.576 |
-0.552 |
-0.142 |
| H7 |
-3.191 |
-1.164 |
-0.219 |
| H8 |
-2.267 |
-0.929 |
1.279 |
| H9 |
-1.498 |
-1.693 |
-0.131 |
| H10 |
-1.715 |
0.475 |
-1.414 |
| H11 |
-2.477 |
1.228 |
-0.011 |
| H12 |
-0.135 |
1.898 |
-0.112 |
| H13 |
-0.375 |
0.852 |
1.322 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5248 | 2.5433 | 2.9537 | 4.1476 | 4.8017 | 1.0976 | 1.0994 | 1.0970 | 2.1685 | 2.1655 | 3.4989 | 2.7830 |
C2 | 1.5248 | | 1.5149 | 2.3839 | 3.6083 | 4.4478 | 2.1713 | 2.1753 | 2.1747 | 1.0996 | 1.1001 | 2.1737 | 2.1753 | C3 | 2.5433 | 1.5149 | | 1.4403 | 2.3002 | 3.3276 | 3.4898 | 2.8080 | 2.8249 | 2.1393 | 2.1154 | 1.0985 | 1.0992 | O4 | 2.9537 | 2.3839 | 1.4403 | | 1.3727 | 2.0670 | 3.9377 | 3.3702 | 2.6636 | 2.6074 | 3.3190 | 2.0678 | 2.0711 | N5 | 4.1476 | 3.6083 | 2.3002 | 1.3727 | | 1.1790 | 5.1945 | 4.3229 | 3.8227 | 3.9188 | 4.4022 | 2.6018 | 2.4777 | O6 | 4.8017 | 4.4478 | 3.3276 | 2.0670 | 1.1790 | | 5.8004 | 5.0611 | 4.2310 | 4.5922 | 5.3588 | 3.6542 | 3.5813 | H7 | 1.0976 | 2.1713 | 3.4898 | 3.9377 | 5.1945 | 5.8004 | | 1.7758 | 1.7763 | 2.5084 | 2.5054 | 4.3272 | 3.7910 | H8 | 1.0994 | 2.1753 | 2.8080 | 3.3702 | 4.3229 | 5.0611 | 1.7758 | | 1.7779 | 3.0865 | 2.5221 | 3.8035 | 2.5988 | H9 | 1.0970 | 2.1747 | 2.8249 | 2.6636 | 3.8227 | 4.2310 | 1.7763 | 1.7779 | | 2.5287 | 3.0833 | 3.8407 | 3.1386 | H10 | 2.1685 | 1.0996 | 2.1393 | 2.6074 | 3.9188 | 4.5922 | 2.5084 | 3.0865 | 2.5287 | | 1.7655 | 2.4935 | 3.0703 | H11 | 2.1655 | 1.1001 | 2.1154 | 3.3190 | 4.4022 | 5.3588 | 2.5054 | 2.5221 | 3.0833 | 1.7655 | | 2.4372 | 2.5169 | H12 | 3.4989 | 2.1737 | 1.0985 | 2.0678 | 2.6018 | 3.6542 | 4.3272 | 3.8035 | 3.8407 | 2.4935 | 2.4372 | | 1.7907 | H13 | 2.7830 | 2.1753 | 1.0992 | 2.0711 | 2.4777 | 3.5813 | 3.7910 | 2.5988 | 3.1386 | 3.0703 | 2.5169 | 1.7907 | |
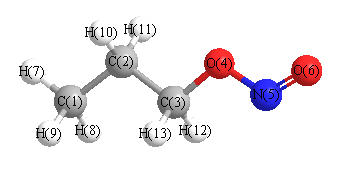 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
113.584 |
|
C1 |
C2 |
H10 |
110.390 |
| C1 |
C2 |
H11 |
110.127 |
|
C2 |
C1 |
H7 |
110.729 |
| C2 |
C1 |
H8 |
110.944 |
|
C2 |
C1 |
H9 |
111.037 |
| C2 |
C3 |
O4 |
107.520 |
|
C2 |
C3 |
H12 |
111.567 |
| C2 |
C3 |
H13 |
111.646 |
|
C3 |
C2 |
H10 |
108.778 |
| C3 |
C2 |
H11 |
106.920 |
|
C3 |
O4 |
N5 |
109.691 |
| O4 |
C3 |
H12 |
108.325 |
|
O4 |
C3 |
H13 |
108.542 |
| O4 |
N5 |
O6 |
107.960 |
|
H7 |
C1 |
H8 |
107.855 |
| H7 |
C1 |
H9 |
108.067 |
|
H8 |
C1 |
H9 |
108.084 |
| H10 |
C2 |
H11 |
106.764 |
|
H12 |
C3 |
H13 |
109.130 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.480 |
|
|
|
| 2 |
C |
0.476 |
|
|
|
| 3 |
C |
0.902 |
|
|
|
| 4 |
O |
-0.559 |
|
|
|
| 5 |
N |
0.490 |
|
|
|
| 6 |
O |
-0.478 |
|
|
|
| 7 |
H |
-0.153 |
|
|
|
| 8 |
H |
-0.137 |
|
|
|
| 9 |
H |
-0.118 |
|
|
|
| 10 |
H |
-0.236 |
|
|
|
| 11 |
H |
-0.174 |
|
|
|
| 12 |
H |
-0.314 |
|
|
|
| 13 |
H |
-0.180 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.648 |
-0.947 |
0.713 |
2.902 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.251 |
0.525 |
-0.678 |
| y |
0.525 |
-35.187 |
-0.877 |
| z |
-0.678 |
-0.877 |
-37.313 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.001 |
0.525 |
-0.678 |
| y |
0.525 |
3.095 |
-0.877 |
| z |
-0.678 |
-0.877 |
-0.094 |
|
| Polar |
|---|
| 3z2-r2 | -0.188 |
|---|
| x2-y2 | -4.064 |
|---|
| xy | 0.525 |
|---|
| xz | -0.678 |
|---|
| yz | -0.877 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.398 |
-0.981 |
0.001 |
| y |
-0.981 |
8.109 |
0.015 |
| z |
0.001 |
0.015 |
7.160 |
<r2> (average value of r
2) Å
2
| <r2> |
193.992 |
| (<r2>)1/2 |
13.928 |