Vibrational Frequencies calculated at PBE1PBE/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3150 |
3029 |
26.61 |
|
|
|
| 2 |
A' |
3140 |
3019 |
29.49 |
|
|
|
| 3 |
A' |
3065 |
2947 |
25.58 |
|
|
|
| 4 |
A' |
3049 |
2931 |
28.22 |
|
|
|
| 5 |
A' |
2992 |
2877 |
81.06 |
|
|
|
| 6 |
A' |
2965 |
2852 |
44.55 |
|
|
|
| 7 |
A' |
1497 |
1440 |
6.56 |
|
|
|
| 8 |
A' |
1480 |
1423 |
4.83 |
|
|
|
| 9 |
A' |
1473 |
1416 |
6.54 |
|
|
|
| 10 |
A' |
1466 |
1409 |
2.51 |
|
|
|
| 11 |
A' |
1451 |
1395 |
0.49 |
|
|
|
| 12 |
A' |
1410 |
1356 |
12.65 |
|
|
|
| 13 |
A' |
1391 |
1338 |
4.54 |
|
|
|
| 14 |
A' |
1313 |
1263 |
2.96 |
|
|
|
| 15 |
A' |
1228 |
1180 |
62.44 |
|
|
|
| 16 |
A' |
1191 |
1145 |
124.32 |
|
|
|
| 17 |
A' |
1128 |
1085 |
9.10 |
|
|
|
| 18 |
A' |
1080 |
1039 |
3.08 |
|
|
|
| 19 |
A' |
1008 |
969 |
22.72 |
|
|
|
| 20 |
A' |
911 |
876 |
6.30 |
|
|
|
| 21 |
A' |
442 |
425 |
0.67 |
|
|
|
| 22 |
A' |
410 |
394 |
2.87 |
|
|
|
| 23 |
A' |
193 |
186 |
1.28 |
|
|
|
| 24 |
A" |
3131 |
3011 |
56.04 |
|
|
|
| 25 |
A" |
3106 |
2986 |
2.05 |
|
|
|
| 26 |
A" |
3049 |
2932 |
54.35 |
|
|
|
| 27 |
A" |
3000 |
2885 |
57.32 |
|
|
|
| 28 |
A" |
1472 |
1415 |
7.43 |
|
|
|
| 29 |
A" |
1458 |
1402 |
8.50 |
|
|
|
| 30 |
A" |
1300 |
1250 |
0.28 |
|
|
|
| 31 |
A" |
1254 |
1206 |
1.85 |
|
|
|
| 32 |
A" |
1183 |
1138 |
4.50 |
|
|
|
| 33 |
A" |
1156 |
1111 |
0.22 |
|
|
|
| 34 |
A" |
890 |
856 |
1.29 |
|
|
|
| 35 |
A" |
755 |
726 |
1.81 |
|
|
|
| 36 |
A" |
238 |
229 |
0.92 |
|
|
|
| 37 |
A" |
226 |
218 |
2.38 |
|
|
|
| 38 |
A" |
108 |
104 |
2.88 |
|
|
|
| 39 |
A" |
105 |
101 |
0.13 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29930.1 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 28780.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
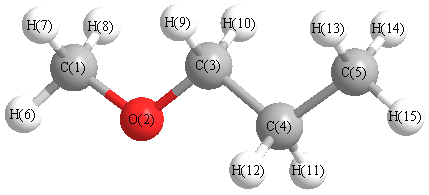
Charges from optimized geometry at PBE1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.732 |
|
|
|
| 2 |
O |
-0.580 |
|
|
|
| 3 |
C |
0.846 |
|
|
|
| 4 |
C |
0.514 |
|
|
|
| 5 |
C |
0.424 |
|
|
|
| 6 |
H |
-0.317 |
|
|
|
| 7 |
H |
-0.088 |
|
|
|
| 8 |
H |
-0.088 |
|
|
|
| 9 |
H |
-0.296 |
|
|
|
| 10 |
H |
-0.296 |
|
|
|
| 11 |
H |
-0.227 |
|
|
|
| 12 |
H |
-0.227 |
|
|
|
| 13 |
H |
-0.124 |
|
|
|
| 14 |
H |
-0.124 |
|
|
|
| 15 |
H |
-0.149 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.294 |
1.023 |
0.000 |
1.064 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.733 |
-2.161 |
0.000 |
| y |
-2.161 |
-33.991 |
0.000 |
| z |
0.000 |
0.000 |
-33.236 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.880 |
-2.161 |
0.000 |
| y |
-2.161 |
-1.506 |
0.000 |
| z |
0.000 |
0.000 |
-0.374 |
|
| Polar |
|---|
| 3z2-r2 | -0.748 |
|---|
| x2-y2 | 2.258 |
|---|
| xy | -2.161 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.366 |
-0.447 |
0.000 |
| y |
-0.447 |
8.200 |
0.000 |
| z |
0.000 |
0.000 |
7.685 |
<r2> (average value of r
2) Å
2
| <r2> |
180.571 |
| (<r2>)1/2 |
13.438 |