Vibrational Frequencies calculated at PBE1PBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3247 |
3119 |
9.42 |
|
|
|
| 2 |
A' |
3240 |
3112 |
5.61 |
|
|
|
| 3 |
A' |
3232 |
3104 |
8.62 |
|
|
|
| 4 |
A' |
3223 |
3095 |
4.05 |
|
|
|
| 5 |
A' |
3214 |
3086 |
0.04 |
|
|
|
| 6 |
A' |
3192 |
3065 |
7.50 |
|
|
|
| 7 |
A' |
3074 |
2952 |
1.18 |
|
|
|
| 8 |
A' |
1751 |
1681 |
93.01 |
|
|
|
| 9 |
A' |
1655 |
1590 |
15.63 |
|
|
|
| 10 |
A' |
1635 |
1571 |
8.87 |
|
|
|
| 11 |
A' |
1548 |
1487 |
0.31 |
|
|
|
| 12 |
A' |
1519 |
1459 |
14.18 |
|
|
|
| 13 |
A' |
1510 |
1450 |
19.90 |
|
|
|
| 14 |
A' |
1427 |
1370 |
54.43 |
|
|
|
| 15 |
A' |
1384 |
1329 |
4.65 |
|
|
|
| 16 |
A' |
1361 |
1307 |
1.92 |
|
|
|
| 17 |
A' |
1295 |
1244 |
189.86 |
|
|
|
| 18 |
A' |
1240 |
1191 |
21.69 |
|
|
|
| 19 |
A' |
1230 |
1181 |
8.56 |
|
|
|
| 20 |
A' |
1130 |
1085 |
9.59 |
|
|
|
| 21 |
A' |
1122 |
1077 |
1.75 |
|
|
|
| 22 |
A' |
1067 |
1025 |
8.07 |
|
|
|
| 23 |
A' |
1046 |
1005 |
0.00 |
|
|
|
| 24 |
A' |
976 |
938 |
27.45 |
|
|
|
| 25 |
A' |
758 |
728 |
0.10 |
|
|
|
| 26 |
A' |
657 |
631 |
0.67 |
|
|
|
| 27 |
A' |
595 |
571 |
21.88 |
|
|
|
| 28 |
A' |
479 |
460 |
0.68 |
|
|
|
| 29 |
A' |
375 |
361 |
1.46 |
|
|
|
| 30 |
A' |
225 |
216 |
5.42 |
|
|
|
| 31 |
A" |
3142 |
3018 |
3.43 |
|
|
|
| 32 |
A" |
1542 |
1481 |
16.48 |
|
|
|
| 33 |
A" |
1097 |
1054 |
3.10 |
|
|
|
| 34 |
A" |
1058 |
1016 |
0.94 |
|
|
|
| 35 |
A" |
1034 |
993 |
0.73 |
|
|
|
| 36 |
A" |
978 |
940 |
3.99 |
|
|
|
| 37 |
A" |
889 |
853 |
0.35 |
|
|
|
| 38 |
A" |
814 |
782 |
26.35 |
|
|
|
| 39 |
A" |
738 |
708 |
70.59 |
|
|
|
| 40 |
A" |
638 |
613 |
12.82 |
|
|
|
| 41 |
A" |
442 |
424 |
1.39 |
|
|
|
| 42 |
A" |
429 |
412 |
0.02 |
|
|
|
| 43 |
A" |
204 |
196 |
1.05 |
|
|
|
| 44 |
A" |
160 |
154 |
0.03 |
|
|
|
| 45 |
A" |
84 |
81 |
3.09 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30827.0 cm
-1
Scaled (by 0.9604) Zero Point Vibrational Energy (zpe) 29606.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
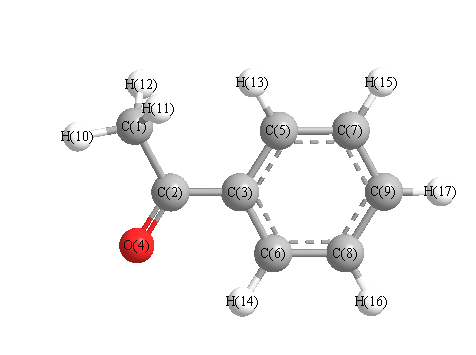
Charges from optimized geometry at PBE1PBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.713 |
|
|
|
| 2 |
C |
0.441 |
|
|
|
| 3 |
C |
-0.119 |
|
|
|
| 4 |
O |
-0.465 |
|
|
|
| 5 |
C |
-0.224 |
|
|
|
| 6 |
C |
-0.180 |
|
|
|
| 7 |
C |
-0.218 |
|
|
|
| 8 |
C |
-0.224 |
|
|
|
| 9 |
C |
-0.213 |
|
|
|
| 10 |
H |
0.257 |
|
|
|
| 11 |
H |
0.245 |
|
|
|
| 12 |
H |
0.245 |
|
|
|
| 13 |
H |
0.225 |
|
|
|
| 14 |
H |
0.254 |
|
|
|
| 15 |
H |
0.228 |
|
|
|
| 16 |
H |
0.230 |
|
|
|
| 17 |
H |
0.231 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.010 |
-2.120 |
0.000 |
2.921 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.135 |
5.682 |
0.000 |
| y |
5.682 |
-51.685 |
0.000 |
| z |
0.000 |
0.000 |
-56.057 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.736 |
5.682 |
0.000 |
| y |
5.682 |
-0.089 |
0.000 |
| z |
0.000 |
0.000 |
-6.647 |
|
| Polar |
|---|
| 3z2-r2 | -13.294 |
|---|
| x2-y2 | 4.550 |
|---|
| xy | 5.682 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.768 |
0.368 |
0.000 |
| y |
0.368 |
15.688 |
0.000 |
| z |
0.000 |
0.000 |
4.250 |
<r2> (average value of r
2) Å
2
| <r2> |
343.187 |
| (<r2>)1/2 |
18.525 |