Jump to
S1C2
Vibrational Frequencies calculated at PBE1PBE/6-311G**
Geometric Data calculated at PBE1PBE/6-311G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at PBE1PBE/6-311G**
| | hartrees |
|---|
| Energy at 0K | -235.609651 |
| Energy at 298.15K | -235.623055 |
| HF Energy | -235.609651 |
| Nuclear repulsion energy | 250.180245 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBE1PBE/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3117 |
2991 |
114.06 |
|
|
|
| 2 |
A' |
3110 |
2984 |
16.53 |
|
|
|
| 3 |
A' |
3106 |
2980 |
27.79 |
|
|
|
| 4 |
A' |
3072 |
2947 |
18.79 |
|
|
|
| 5 |
A' |
3059 |
2935 |
10.52 |
|
|
|
| 6 |
A' |
3053 |
2929 |
5.34 |
|
|
|
| 7 |
A' |
3035 |
2912 |
30.48 |
|
|
|
| 8 |
A' |
3034 |
2911 |
28.38 |
|
|
|
| 9 |
A' |
1498 |
1437 |
10.96 |
|
|
|
| 10 |
A' |
1491 |
1430 |
3.32 |
|
|
|
| 11 |
A' |
1487 |
1426 |
2.70 |
|
|
|
| 12 |
A' |
1407 |
1350 |
9.85 |
|
|
|
| 13 |
A' |
1406 |
1349 |
5.14 |
|
|
|
| 14 |
A' |
1379 |
1323 |
0.29 |
|
|
|
| 15 |
A' |
1363 |
1308 |
8.01 |
|
|
|
| 16 |
A' |
1278 |
1226 |
4.44 |
|
|
|
| 17 |
A' |
1254 |
1203 |
0.54 |
|
|
|
| 18 |
A' |
1201 |
1152 |
0.25 |
|
|
|
| 19 |
A' |
1151 |
1104 |
0.22 |
|
|
|
| 20 |
A' |
1057 |
1014 |
0.25 |
|
|
|
| 21 |
A' |
961 |
922 |
1.12 |
|
|
|
| 22 |
A' |
951 |
913 |
0.87 |
|
|
|
| 23 |
A' |
873 |
838 |
1.27 |
|
|
|
| 24 |
A' |
816 |
783 |
1.37 |
|
|
|
| 25 |
A' |
649 |
623 |
1.82 |
|
|
|
| 26 |
A' |
425 |
408 |
0.13 |
|
|
|
| 27 |
A' |
326 |
313 |
0.11 |
|
|
|
| 28 |
A' |
97 |
93 |
0.01 |
|
|
|
| 29 |
A" |
3114 |
2987 |
36.43 |
|
|
|
| 30 |
A" |
3111 |
2985 |
26.45 |
|
|
|
| 31 |
A" |
3108 |
2982 |
9.83 |
|
|
|
| 32 |
A" |
3052 |
2928 |
71.62 |
|
|
|
| 33 |
A" |
1492 |
1432 |
5.83 |
|
|
|
| 34 |
A" |
1491 |
1430 |
0.28 |
|
|
|
| 35 |
A" |
1463 |
1404 |
6.77 |
|
|
|
| 36 |
A" |
1292 |
1239 |
0.24 |
|
|
|
| 37 |
A" |
1271 |
1220 |
0.00 |
|
|
|
| 38 |
A" |
1228 |
1178 |
0.08 |
|
|
|
| 39 |
A" |
1144 |
1098 |
0.61 |
|
|
|
| 40 |
A" |
1132 |
1086 |
0.07 |
|
|
|
| 41 |
A" |
996 |
956 |
4.20 |
|
|
|
| 42 |
A" |
922 |
884 |
0.00 |
|
|
|
| 43 |
A" |
912 |
875 |
0.00 |
|
|
|
| 44 |
A" |
806 |
773 |
0.25 |
|
|
|
| 45 |
A" |
374 |
359 |
0.01 |
|
|
|
| 46 |
A" |
261 |
250 |
0.12 |
|
|
|
| 47 |
A" |
253 |
243 |
0.00 |
|
|
|
| 48 |
A" |
234 |
225 |
0.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36657.3 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 35169.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBE1PBE/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.256 |
0.085 |
1.078 |
| C2 |
0.256 |
0.085 |
-1.078 |
| C3 |
-0.660 |
0.710 |
0.000 |
| C4 |
0.850 |
-0.854 |
0.000 |
| C5 |
-0.861 |
2.211 |
0.000 |
| C6 |
0.256 |
-2.252 |
0.000 |
| H7 |
0.993 |
0.807 |
1.445 |
| H8 |
-0.227 |
-0.388 |
1.938 |
| H9 |
0.993 |
0.807 |
-1.445 |
| H10 |
-0.227 |
-0.388 |
-1.938 |
| H11 |
-1.634 |
0.210 |
0.000 |
| H12 |
1.942 |
-0.922 |
0.000 |
| H13 |
0.103 |
2.732 |
0.000 |
| H14 |
-0.839 |
-2.219 |
0.000 |
| H15 |
-1.418 |
2.536 |
-0.885 |
| H16 |
-1.418 |
2.536 |
0.885 |
| H17 |
0.573 |
-2.813 |
-0.886 |
| H18 |
0.573 |
-2.813 |
0.886 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.1558 | 1.5461 | 1.5485 | 2.6321 | 2.5744 | 1.0947 | 1.0942 | 2.7257 | 3.0909 | 2.1794 | 2.2400 | 2.8614 | 2.7694 | 3.5582 | 2.9742 | 3.5150 | 2.9217 |
C2 | 2.1558 | | 1.5461 | 1.5485 | 2.6321 | 2.5744 | 2.7257 | 3.0909 | 1.0947 | 1.0942 | 2.1794 | 2.2400 | 2.8614 | 2.7694 | 2.9742 | 3.5582 | 2.9217 | 3.5150 | C3 | 1.5461 | 1.5461 | | 2.1740 | 1.5146 | 3.1008 | 2.1976 | 2.2690 | 2.1976 | 2.2690 | 1.0951 | 3.0707 | 2.1605 | 2.9342 | 2.1663 | 2.1663 | 3.8357 | 3.8357 | C4 | 1.5485 | 1.5485 | 2.1740 | | 3.5107 | 1.5188 | 2.2066 | 2.2658 | 2.2066 | 2.2658 | 2.7024 | 1.0940 | 3.6630 | 2.1707 | 4.1741 | 4.1741 | 2.1670 | 2.1670 | C5 | 2.6321 | 2.6321 | 1.5146 | 3.5107 | | 4.6013 | 2.7383 | 3.3033 | 2.7383 | 3.3033 | 2.1451 | 4.2035 | 1.0953 | 4.4300 | 1.0950 | 1.0950 | 5.2989 | 5.2989 | C6 | 2.5744 | 2.5744 | 3.1008 | 1.5188 | 4.6013 | | 3.4627 | 2.7326 | 3.4627 | 2.7326 | 3.1044 | 2.1475 | 4.9864 | 1.0953 | 5.1495 | 5.1495 | 1.0947 | 1.0947 | H7 | 1.0947 | 2.7257 | 2.1976 | 2.2066 | 2.7383 | 3.4627 | | 1.7771 | 2.8905 | 3.7898 | 3.0571 | 2.4446 | 2.5664 | 3.8206 | 3.7727 | 3.0196 | 4.3255 | 3.6865 | H8 | 1.0942 | 3.0909 | 2.2690 | 2.2658 | 3.3033 | 2.7326 | 1.7771 | | 3.7898 | 3.8762 | 2.4684 | 2.9572 | 3.6871 | 2.7355 | 4.2352 | 3.3282 | 3.8071 | 2.7619 | H9 | 2.7257 | 1.0947 | 2.1976 | 2.2066 | 2.7383 | 3.4627 | 2.8905 | 3.7898 | | 1.7771 | 3.0571 | 2.4446 | 2.5664 | 3.8206 | 3.0196 | 3.7727 | 3.6865 | 4.3255 | H10 | 3.0909 | 1.0942 | 2.2690 | 2.2658 | 3.3033 | 2.7326 | 3.7898 | 3.8762 | 1.7771 | | 2.4684 | 2.9572 | 3.6871 | 2.7355 | 3.3282 | 4.2352 | 2.7619 | 3.8071 | H11 | 2.1794 | 2.1794 | 1.0951 | 2.7024 | 2.1451 | 3.1044 | 3.0571 | 2.4684 | 3.0571 | 2.4684 | | 3.7506 | 3.0617 | 2.5558 | 2.4980 | 2.4980 | 3.8460 | 3.8460 | H12 | 2.2400 | 2.2400 | 3.0707 | 1.0940 | 4.2035 | 2.1475 | 2.4446 | 2.9572 | 2.4446 | 2.9572 | 3.7506 | | 4.0900 | 3.0680 | 4.9018 | 4.9018 | 2.4969 | 2.4969 | H13 | 2.8614 | 2.8614 | 2.1605 | 3.6630 | 1.0953 | 4.9864 | 2.5664 | 3.6871 | 2.5664 | 3.6871 | 3.0617 | 4.0900 | | 5.0390 | 1.7703 | 1.7703 | 5.6342 | 5.6342 | H14 | 2.7694 | 2.7694 | 2.9342 | 2.1707 | 4.4300 | 1.0953 | 3.8206 | 2.7355 | 3.8206 | 2.7355 | 2.5558 | 3.0680 | 5.0390 | | 4.8712 | 4.8712 | 1.7689 | 1.7689 | H15 | 3.5582 | 2.9742 | 2.1663 | 4.1741 | 1.0950 | 5.1495 | 3.7727 | 4.2352 | 3.0196 | 3.3282 | 2.4980 | 4.9018 | 1.7703 | 4.8712 | | 1.7697 | 5.7073 | 5.9757 | H16 | 2.9742 | 3.5582 | 2.1663 | 4.1741 | 1.0950 | 5.1495 | 3.0196 | 3.3282 | 3.7727 | 4.2352 | 2.4980 | 4.9018 | 1.7703 | 4.8712 | 1.7697 | | 5.9757 | 5.7073 | H17 | 3.5150 | 2.9217 | 3.8357 | 2.1670 | 5.2989 | 1.0947 | 4.3255 | 3.8071 | 3.6865 | 2.7619 | 3.8460 | 2.4969 | 5.6342 | 1.7689 | 5.7073 | 5.9757 | | 1.7713 | H18 | 2.9217 | 3.5150 | 3.8357 | 2.1670 | 5.2989 | 1.0947 | 3.6865 | 2.7619 | 4.3255 | 3.8071 | 3.8460 | 2.4969 | 5.6342 | 1.7689 | 5.9757 | 5.7073 | 1.7713 | |
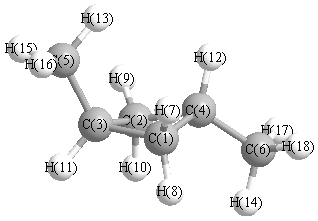 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
88.400 |
|
C1 |
C3 |
C5 |
118.628 |
| C1 |
C3 |
H11 |
110.032 |
|
C1 |
C4 |
C2 |
88.227 |
| C1 |
C4 |
C6 |
114.133 |
|
C1 |
C4 |
H12 |
114.835 |
| C2 |
C3 |
C5 |
118.628 |
|
C2 |
C3 |
H11 |
110.032 |
| C2 |
C4 |
C6 |
114.133 |
|
C2 |
C4 |
H12 |
114.835 |
| C3 |
C1 |
C4 |
89.256 |
|
C3 |
C1 |
H7 |
111.500 |
| C3 |
C1 |
H8 |
117.469 |
|
C3 |
C2 |
C4 |
89.256 |
| C3 |
C2 |
H9 |
111.500 |
|
C3 |
C2 |
H10 |
117.469 |
| C3 |
C5 |
H13 |
110.730 |
|
C3 |
C5 |
H15 |
111.215 |
| C3 |
C5 |
H16 |
111.215 |
|
C4 |
C1 |
H7 |
112.050 |
| C4 |
C1 |
H8 |
117.006 |
|
C4 |
C2 |
H9 |
112.050 |
| C4 |
C2 |
H10 |
117.006 |
|
C4 |
C6 |
H14 |
111.242 |
| C4 |
C6 |
H17 |
110.982 |
|
C4 |
C6 |
H18 |
110.982 |
| C5 |
C3 |
H11 |
109.519 |
|
C6 |
C4 |
H12 |
109.484 |
| H7 |
C1 |
H8 |
108.561 |
|
H9 |
C2 |
H10 |
108.561 |
| H13 |
C5 |
H15 |
107.853 |
|
H13 |
C5 |
H16 |
107.853 |
| H14 |
C6 |
H17 |
107.740 |
|
H14 |
C6 |
H18 |
107.740 |
| H15 |
C5 |
H16 |
107.825 |
|
H17 |
C6 |
H18 |
108.004 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.171 |
|
|
|
| 2 |
C |
-0.171 |
|
|
|
| 3 |
C |
-0.267 |
|
|
|
| 4 |
C |
-0.268 |
|
|
|
| 5 |
C |
-0.287 |
|
|
|
| 6 |
C |
-0.296 |
|
|
|
| 7 |
H |
0.122 |
|
|
|
| 8 |
H |
0.122 |
|
|
|
| 9 |
H |
0.122 |
|
|
|
| 10 |
H |
0.122 |
|
|
|
| 11 |
H |
0.133 |
|
|
|
| 12 |
H |
0.124 |
|
|
|
| 13 |
H |
0.111 |
|
|
|
| 14 |
H |
0.113 |
|
|
|
| 15 |
H |
0.121 |
|
|
|
| 16 |
H |
0.121 |
|
|
|
| 17 |
H |
0.123 |
|
|
|
| 18 |
H |
0.123 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.020 |
-0.005 |
0.000 |
0.020 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.568 |
0.341 |
0.000 |
| y |
0.341 |
-40.610 |
0.000 |
| z |
0.000 |
0.000 |
-40.411 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.942 |
0.341 |
0.000 |
| y |
0.341 |
-0.621 |
0.000 |
| z |
0.000 |
0.000 |
-0.322 |
|
| Polar |
|---|
| 3z2-r2 | -0.643 |
|---|
| x2-y2 | 1.042 |
|---|
| xy | 0.341 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.182 |
-0.686 |
0.000 |
| y |
-0.686 |
11.058 |
0.000 |
| z |
0.000 |
0.000 |
9.580 |
<r2> (average value of r
2) Å
2
| <r2> |
190.875 |
| (<r2>)1/2 |
13.816 |