Vibrational Frequencies calculated at PBE1PBE/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3226 |
3100 |
14.96 |
|
|
|
| 2 |
A' |
3127 |
3005 |
10.15 |
|
|
|
| 3 |
A' |
3125 |
3003 |
53.98 |
|
|
|
| 4 |
A' |
3121 |
2999 |
17.11 |
|
|
|
| 5 |
A' |
3073 |
2953 |
50.53 |
|
|
|
| 6 |
A' |
3058 |
2939 |
38.12 |
|
|
|
| 7 |
A' |
1657 |
1593 |
2.33 |
|
|
|
| 8 |
A' |
1507 |
1448 |
1.30 |
|
|
|
| 9 |
A' |
1480 |
1423 |
5.83 |
|
|
|
| 10 |
A' |
1330 |
1278 |
0.74 |
|
|
|
| 11 |
A' |
1316 |
1265 |
1.66 |
|
|
|
| 12 |
A' |
1192 |
1145 |
0.18 |
|
|
|
| 13 |
A' |
1148 |
1104 |
5.73 |
|
|
|
| 14 |
A' |
1114 |
1071 |
1.53 |
|
|
|
| 15 |
A' |
1041 |
1000 |
1.36 |
|
|
|
| 16 |
A' |
988 |
949 |
0.53 |
|
|
|
| 17 |
A' |
967 |
929 |
0.55 |
|
|
|
| 18 |
A' |
929 |
893 |
0.85 |
|
|
|
| 19 |
A' |
900 |
864 |
6.59 |
|
|
|
| 20 |
A' |
823 |
790 |
0.61 |
|
|
|
| 21 |
A' |
786 |
755 |
6.30 |
|
|
|
| 22 |
A' |
734 |
705 |
41.22 |
|
|
|
| 23 |
A' |
474 |
455 |
1.23 |
|
|
|
| 24 |
A' |
377 |
363 |
2.61 |
|
|
|
| 25 |
A" |
3200 |
3076 |
7.97 |
|
|
|
| 26 |
A" |
3122 |
3000 |
66.48 |
|
|
|
| 27 |
A" |
3107 |
2986 |
3.24 |
|
|
|
| 28 |
A" |
3061 |
2941 |
25.76 |
|
|
|
| 29 |
A" |
1483 |
1425 |
1.39 |
|
|
|
| 30 |
A" |
1372 |
1318 |
10.76 |
|
|
|
| 31 |
A" |
1306 |
1255 |
3.21 |
|
|
|
| 32 |
A" |
1289 |
1239 |
0.12 |
|
|
|
| 33 |
A" |
1281 |
1231 |
1.15 |
|
|
|
| 34 |
A" |
1238 |
1190 |
0.50 |
|
|
|
| 35 |
A" |
1198 |
1151 |
0.17 |
|
|
|
| 36 |
A" |
1140 |
1095 |
0.39 |
|
|
|
| 37 |
A" |
1054 |
1012 |
0.82 |
|
|
|
| 38 |
A" |
972 |
935 |
0.03 |
|
|
|
| 39 |
A" |
968 |
931 |
1.22 |
|
|
|
| 40 |
A" |
937 |
901 |
3.92 |
|
|
|
| 41 |
A" |
855 |
822 |
4.47 |
|
|
|
| 42 |
A" |
812 |
780 |
1.68 |
|
|
|
| 43 |
A" |
680 |
653 |
0.68 |
|
|
|
| 44 |
A" |
488 |
469 |
0.17 |
|
|
|
| 45 |
A" |
253 |
243 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 33653.8 cm
-1
Scaled (by 0.961) Zero Point Vibrational Energy (zpe) 32341.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
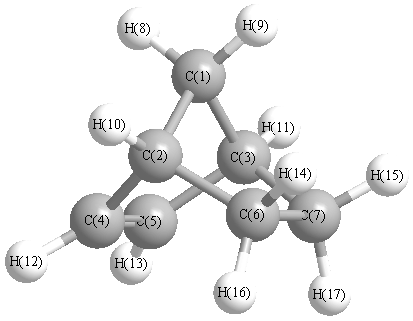
Charges from optimized geometry at PBE1PBE/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.155 |
|
|
|
| 2 |
C |
-0.028 |
|
|
|
| 3 |
C |
-0.028 |
|
|
|
| 4 |
C |
-0.179 |
|
|
|
| 5 |
C |
-0.179 |
|
|
|
| 6 |
C |
-0.177 |
|
|
|
| 7 |
C |
-0.177 |
|
|
|
| 8 |
H |
0.086 |
|
|
|
| 9 |
H |
0.078 |
|
|
|
| 10 |
H |
0.075 |
|
|
|
| 11 |
H |
0.075 |
|
|
|
| 12 |
H |
0.122 |
|
|
|
| 13 |
H |
0.122 |
|
|
|
| 14 |
H |
0.094 |
|
|
|
| 15 |
H |
0.094 |
|
|
|
| 16 |
H |
0.088 |
|
|
|
| 17 |
H |
0.088 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.172 |
-0.298 |
0.000 |
0.345 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.441 |
-0.304 |
0.000 |
| y |
-0.304 |
-45.152 |
0.000 |
| z |
0.000 |
0.000 |
-42.085 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.177 |
-0.304 |
0.000 |
| y |
-0.304 |
-2.889 |
0.000 |
| z |
0.000 |
0.000 |
1.712 |
|
| Polar |
|---|
| 3z2-r2 | 3.424 |
|---|
| x2-y2 | 2.710 |
|---|
| xy | -0.304 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.561 |
-0.579 |
0.000 |
| y |
-0.579 |
10.411 |
0.000 |
| z |
0.000 |
0.000 |
11.407 |
<r2> (average value of r
2) Å
2
| <r2> |
163.458 |
| (<r2>)1/2 |
12.785 |