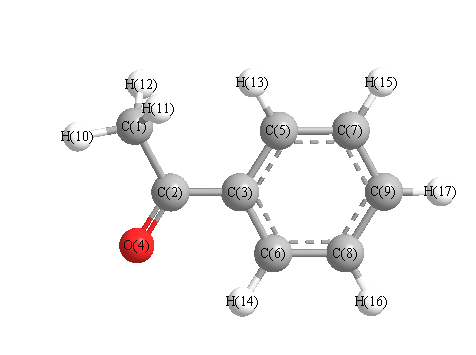
Vibrational Frequencies calculated at PBE1PBE/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3218 |
3093 |
7.77 |
|
|
|
| 2 |
A' |
3213 |
3088 |
9.06 |
|
|
|
| 3 |
A' |
3204 |
3079 |
12.68 |
|
|
|
| 4 |
A' |
3195 |
3071 |
5.09 |
|
|
|
| 5 |
A' |
3185 |
3061 |
0.11 |
|
|
|
| 6 |
A' |
3172 |
3049 |
9.17 |
|
|
|
| 7 |
A' |
3054 |
2935 |
1.96 |
|
|
|
| 8 |
A' |
1794 |
1724 |
184.46 |
|
|
|
| 9 |
A' |
1667 |
1602 |
20.42 |
|
|
|
| 10 |
A' |
1649 |
1585 |
7.74 |
|
|
|
| 11 |
A' |
1533 |
1473 |
0.91 |
|
|
|
| 12 |
A' |
1490 |
1432 |
19.66 |
|
|
|
| 13 |
A' |
1460 |
1403 |
10.01 |
|
|
|
| 14 |
A' |
1381 |
1327 |
80.06 |
|
|
|
| 15 |
A' |
1376 |
1322 |
6.38 |
|
|
|
| 16 |
A' |
1342 |
1289 |
1.86 |
|
|
|
| 17 |
A' |
1294 |
1244 |
158.21 |
|
|
|
| 18 |
A' |
1200 |
1153 |
14.21 |
|
|
|
| 19 |
A' |
1181 |
1135 |
0.48 |
|
|
|
| 20 |
A' |
1112 |
1069 |
7.11 |
|
|
|
| 21 |
A' |
1101 |
1058 |
1.66 |
|
|
|
| 22 |
A' |
1057 |
1016 |
6.57 |
|
|
|
| 23 |
A' |
1025 |
985 |
0.87 |
|
|
|
| 24 |
A' |
963 |
926 |
24.87 |
|
|
|
| 25 |
A' |
749 |
719 |
1.06 |
|
|
|
| 26 |
A' |
628 |
604 |
0.90 |
|
|
|
| 27 |
A' |
599 |
575 |
25.99 |
|
|
|
| 28 |
A' |
466 |
448 |
0.78 |
|
|
|
| 29 |
A' |
366 |
352 |
0.87 |
|
|
|
| 30 |
A' |
215 |
207 |
4.97 |
|
|
|
| 31 |
A" |
3121 |
2999 |
5.05 |
|
|
|
| 32 |
A" |
1470 |
1413 |
10.87 |
|
|
|
| 33 |
A" |
1047 |
1006 |
0.51 |
|
|
|
| 34 |
A" |
1026 |
986 |
0.38 |
|
|
|
| 35 |
A" |
1009 |
969 |
0.18 |
|
|
|
| 36 |
A" |
959 |
922 |
2.57 |
|
|
|
| 37 |
A" |
874 |
840 |
0.16 |
|
|
|
| 38 |
A" |
784 |
753 |
33.53 |
|
|
|
| 39 |
A" |
714 |
687 |
38.02 |
|
|
|
| 40 |
A" |
603 |
580 |
10.93 |
|
|
|
| 41 |
A" |
431 |
414 |
0.32 |
|
|
|
| 42 |
A" |
415 |
399 |
0.01 |
|
|
|
| 43 |
A" |
160 |
154 |
0.17 |
|
|
|
| 44 |
A" |
154 |
148 |
0.00 |
|
|
|
| 45 |
A" |
59 |
57 |
3.51 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30357.6 cm
-1
Scaled (by 0.961) Zero Point Vibrational Energy (zpe) 29173.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.313 |
|
|
|
| 2 |
C |
0.233 |
|
|
|
| 3 |
C |
-0.013 |
|
|
|
| 4 |
O |
-0.279 |
|
|
|
| 5 |
C |
-0.125 |
|
|
|
| 6 |
C |
-0.106 |
|
|
|
| 7 |
C |
-0.125 |
|
|
|
| 8 |
C |
-0.119 |
|
|
|
| 9 |
C |
-0.107 |
|
|
|
| 10 |
H |
0.120 |
|
|
|
| 11 |
H |
0.108 |
|
|
|
| 12 |
H |
0.108 |
|
|
|
| 13 |
H |
0.113 |
|
|
|
| 14 |
H |
0.134 |
|
|
|
| 15 |
H |
0.120 |
|
|
|
| 16 |
H |
0.124 |
|
|
|
| 17 |
H |
0.125 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.855 |
-2.396 |
0.000 |
3.030 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.156 |
5.453 |
0.000 |
| y |
5.453 |
-53.755 |
0.000 |
| z |
0.000 |
0.000 |
-55.012 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
7.228 |
5.453 |
0.000 |
| y |
5.453 |
-2.671 |
0.000 |
| z |
0.000 |
0.000 |
-4.557 |
|
| Polar |
|---|
| 3z2-r2 | -9.114 |
|---|
| x2-y2 | 6.599 |
|---|
| xy | 5.453 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.862 |
0.862 |
0.000 |
| y |
0.862 |
17.996 |
0.000 |
| z |
0.000 |
0.000 |
7.323 |
<r2> (average value of r
2) Å
2
| <r2> |
342.421 |
| (<r2>)1/2 |
18.505 |