Vibrational Frequencies calculated at PBE1PBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3221 |
3087 |
0.00 |
|
|
|
| 2 |
Ag |
3204 |
3072 |
0.00 |
|
|
|
| 3 |
Ag |
1744 |
1672 |
0.00 |
|
|
|
| 4 |
Ag |
1542 |
1478 |
0.00 |
|
|
|
| 5 |
Ag |
1448 |
1388 |
0.00 |
|
|
|
| 6 |
Ag |
1198 |
1148 |
0.00 |
|
|
|
| 7 |
Ag |
1160 |
1112 |
0.00 |
|
|
|
| 8 |
Ag |
1029 |
986 |
0.00 |
|
|
|
| 9 |
Ag |
788 |
756 |
0.00 |
|
|
|
| 10 |
Ag |
404 |
388 |
0.00 |
|
|
|
| 11 |
Au |
974 |
933 |
0.00 |
|
|
|
| 12 |
Au |
896 |
858 |
0.00 |
|
|
|
| 13 |
Au |
805 |
771 |
0.00 |
|
|
|
| 14 |
Au |
588 |
564 |
0.00 |
|
|
|
| 15 |
Au |
147 |
141 |
0.00 |
|
|
|
| 16 |
B1g |
974 |
934 |
0.00 |
|
|
|
| 17 |
B1g |
883 |
846 |
0.00 |
|
|
|
| 18 |
B1g |
723 |
693 |
0.00 |
|
|
|
| 19 |
B1g |
463 |
444 |
0.00 |
|
|
|
| 20 |
B1u |
3220 |
3086 |
47.84 |
|
|
|
| 21 |
B1u |
3204 |
3071 |
18.34 |
|
|
|
| 22 |
B1u |
1665 |
1596 |
0.41 |
|
|
|
| 23 |
B1u |
1477 |
1416 |
38.63 |
|
|
|
| 24 |
B1u |
1332 |
1276 |
32.88 |
|
|
|
| 25 |
B1u |
1188 |
1139 |
9.41 |
|
|
|
| 26 |
B1u |
1058 |
1014 |
0.18 |
|
|
|
| 27 |
B1u |
1009 |
967 |
22.80 |
|
|
|
| 28 |
B1u |
627 |
601 |
4.50 |
|
|
|
| 29 |
B2g |
927 |
889 |
0.00 |
|
|
|
| 30 |
B2g |
758 |
726 |
0.00 |
|
|
|
| 31 |
B2g |
432 |
414 |
0.00 |
|
|
|
| 32 |
B2g |
321 |
308 |
0.00 |
|
|
|
| 33 |
B2u |
3215 |
3081 |
57.11 |
|
|
|
| 34 |
B2u |
3191 |
3059 |
0.45 |
|
|
|
| 35 |
B2u |
1667 |
1598 |
1.93 |
|
|
|
| 36 |
B2u |
1495 |
1433 |
6.86 |
|
|
|
| 37 |
B2u |
1303 |
1249 |
4.77 |
|
|
|
| 38 |
B2u |
1160 |
1112 |
12.97 |
|
|
|
| 39 |
B2u |
1081 |
1036 |
0.53 |
|
|
|
| 40 |
B2u |
740 |
709 |
0.54 |
|
|
|
| 41 |
B2u |
216 |
207 |
0.87 |
|
|
|
| 42 |
B3g |
3214 |
3080 |
0.00 |
|
|
|
| 43 |
B3g |
3191 |
3059 |
0.00 |
|
|
|
| 44 |
B3g |
1673 |
1603 |
0.00 |
|
|
|
| 45 |
B3g |
1496 |
1434 |
0.00 |
|
|
|
| 46 |
B3g |
1316 |
1261 |
0.00 |
|
|
|
| 47 |
B3g |
1123 |
1076 |
0.00 |
|
|
|
| 48 |
B3g |
1004 |
962 |
0.00 |
|
|
|
| 49 |
B3g |
612 |
587 |
0.00 |
|
|
|
| 50 |
B3g |
570 |
546 |
0.00 |
|
|
|
| 51 |
B3u |
923 |
885 |
6.29 |
|
|
|
| 52 |
B3u |
746 |
715 |
156.48 |
|
|
|
| 53 |
B3u |
376 |
361 |
3.34 |
|
|
|
| 54 |
B3u |
105 |
101 |
3.68 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34911.0 cm
-1
Scaled (by 0.9585) Zero Point Vibrational Energy (zpe) 33462.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
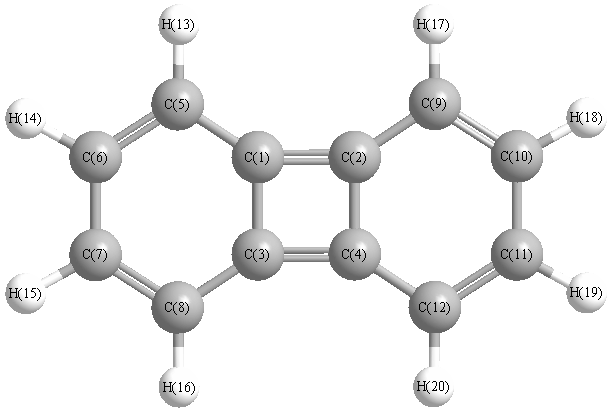
Charges from optimized geometry at PBE1PBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.006 |
|
|
|
| 2 |
C |
0.006 |
|
|
|
| 3 |
C |
0.006 |
|
|
|
| 4 |
C |
0.006 |
|
|
|
| 5 |
C |
-0.207 |
|
|
|
| 6 |
C |
-0.212 |
|
|
|
| 7 |
C |
-0.212 |
|
|
|
| 8 |
C |
-0.207 |
|
|
|
| 9 |
C |
-0.207 |
|
|
|
| 10 |
C |
-0.212 |
|
|
|
| 11 |
C |
-0.212 |
|
|
|
| 12 |
C |
-0.207 |
|
|
|
| 13 |
H |
0.207 |
|
|
|
| 14 |
H |
0.206 |
|
|
|
| 15 |
H |
0.206 |
|
|
|
| 16 |
H |
0.207 |
|
|
|
| 17 |
H |
0.207 |
|
|
|
| 18 |
H |
0.206 |
|
|
|
| 19 |
H |
0.206 |
|
|
|
| 20 |
H |
0.207 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-75.259 |
0.000 |
0.000 |
| y |
0.000 |
-59.481 |
0.000 |
| z |
0.000 |
0.000 |
-58.766 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-16.136 |
0.000 |
0.000 |
| y |
0.000 |
7.532 |
0.000 |
| z |
0.000 |
0.000 |
8.604 |
|
| Polar |
|---|
| 3z2-r2 | 17.208 |
|---|
| x2-y2 | -15.779 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.301 |
0.000 |
0.000 |
| y |
0.000 |
19.148 |
0.000 |
| z |
0.000 |
0.000 |
31.272 |
<r2> (average value of r
2) Å
2
| <r2> |
558.218 |
| (<r2>)1/2 |
23.627 |